PUBLICATIONS
2026
52. Parallel Anti-Sense Concurrent One-Pot/Two-Step Cascade for the Asymmetric Biocatalytic Amination of Racemic sec-Alcohols
A. Rudzka, T. Reiter, W. Kroutil, P. Borowiecki*
ChemCatChem 2026, 11, e70763.
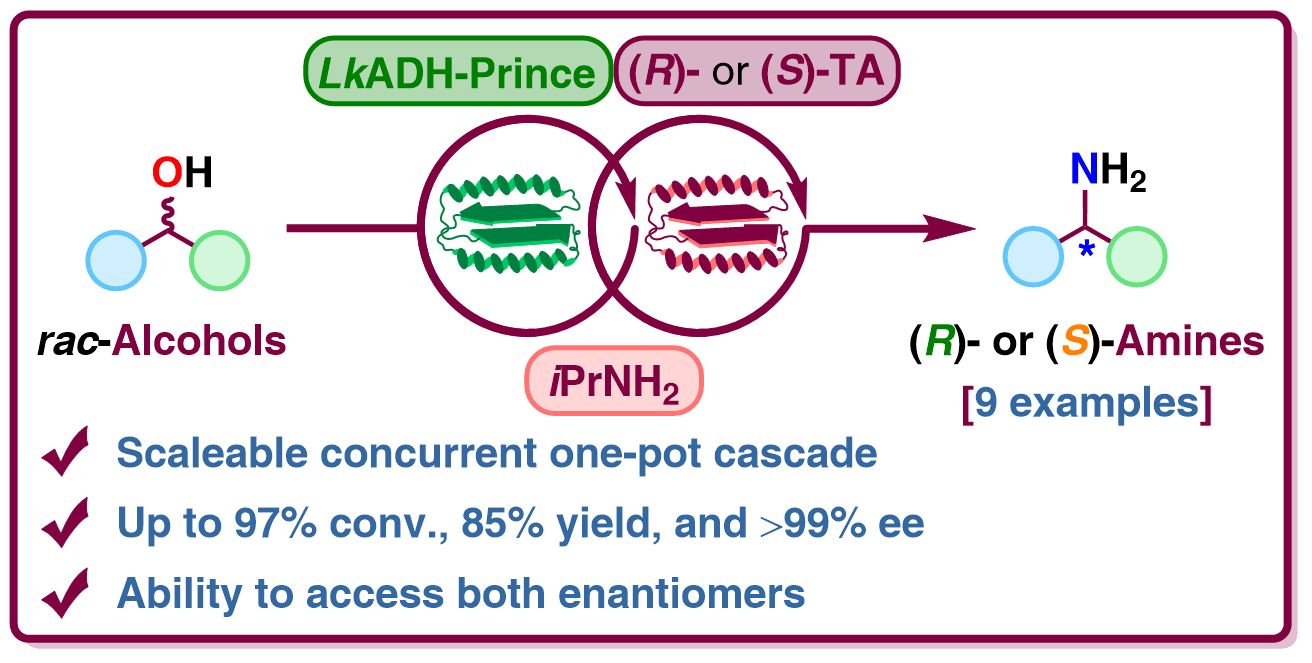
We report the development of a concurrent one-pot/twostep biocatalytic cascade for the asymmetric synthesis of enantiomerically enriched α-chiral primary amines from racemic arylring-containing secondary alcohols via alcohol oxidation employing a non-enantioselective alcohol dehydrogenase, followed by asymmetric reductive amination using a stereoselective transaminase. The amine donor 2-propylamine served, on the one hand, as the nitrogen source for the amination step and, on the other hand, its deaminated/oxidized form (acetone) acted as the oxidant for the first step. The elaborated protocol employed lyophilized Escherichia coli whole cells containing a heterologously expressed ambidextrous alcohol dehydrogenase deduced from Lactobacillus kefir (E. coli/LkADH-Prince) in combination with stereocomplementary recombinant transaminases (E. coli/TAs) that simultaneously converted the in situ-generated carbonyl intermediates into the corresponding optically active amines with high-to-excellent stereocontrol. The developed cascade enables the efficient preparation of nonracemic amines with high conversions (up to 94%), very good isolated yields (up to 85%), and excellent enantiomeric excesses (up to >99%), providing access to both enantiomers under mild, non-toxic, and exclusively aqueous reaction conditions.
doi: 10.1002/cctc.70763
51. Stereoconvergent Photo-Biocatalytic Cascade to Optically Enriched Amines from Racemic Carboxylic Acids
A. Rudzka, A. Madej, T. Reiter, W. Kroutil, P. Borowiecki*
ACS Omega 2026, 4, 6207–6226.
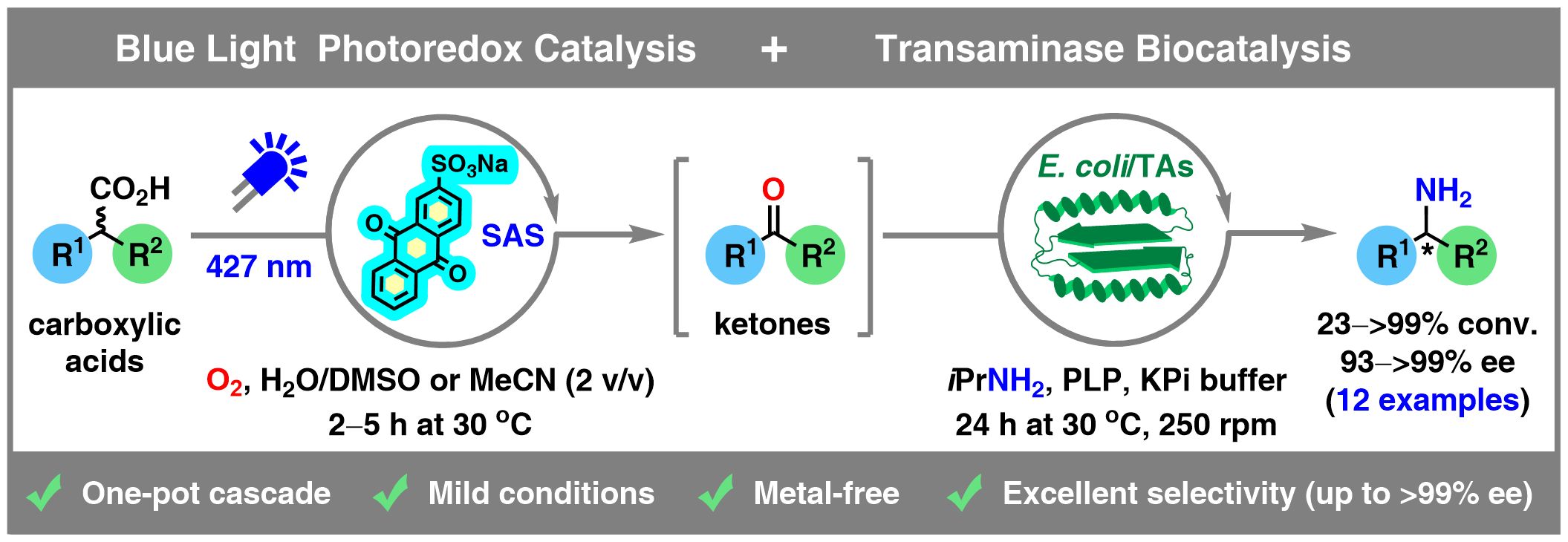
Herein, we report a synergistic synthetic strategy to integrate photoredox catalysis and biocatalysis in a one-pot/two-step linear cascade synthesis of enantiomerically pure amines starting from racemic carboxylic acids utilizing visible light and transaminases. This transformation was achieved via quantitative decarboxylative oxidation (>99% conv.) of racemic aryl-alkyl carboxylic acids followed by stereoselective reductive amination of the corresponding dehomologated carbonyl intermediates. The initial photocatalytic step employs blue LED irradiation (427 nm) and molecular oxygen (O2) as the terminal oxidant in the presence of 2.5 mol % of sodium anthraquinone-2-sulfonate (SAS) as a water-soluble, metal-free photocatalyst, operating in an aqueous medium containing 2% v/v of acetonitrile (MeCN) or dimethyl sulfoxide (DMSO) as the respective cosolvents. The subsequent transaminase-catalyzed reductive amination of the in situ-generated ketones furnished the desired optically active amines in a stereocomplementary manner with enantiomeric excesses ranging from 93% to 99.9% and up to >99% conversions after two steps. Among the tested substrates, three representative nonsteroidal anti-inflammatory drugs (NSAIDs), including ibuprofen, flurbiprofen, and naproxen, were successfully transformed into dehomologated NSAID-derived nonracemic amines with 62–>99% ee, respectively. Furthermore, upscaling of the photobiocatalytic process employing 2-(naphthalen-1-yl)propanoic acid (1.0 mmol) as the substrate enabled the synthesis of (R)-1-(naphthalen-1-yl)ethan-1-amine with 92% product formation and >99% ee. Finally, a one-step transformation of the aforementioned amine using a recently developed betaine-based Mitsunobu reagent furnished the enantiomerically pure calcimimetic drug cinacalcet (>99% ee) in 76% isolated yield, without the need for transition metal catalysts and/or hazardous and flammable hydrogen gas.
50. Photo-Biocatalytic One-Pot Conversion of 1,1-Disubstituted Alkenes into Optically Enriched Primary Amines
N. Antos, J. Skrzypek, P. Wieczorkiewicz, T. Reiter, W. Kroutil, P. Borowiecki*
Adv. Synth. Catal. 2026, 1, e70219.
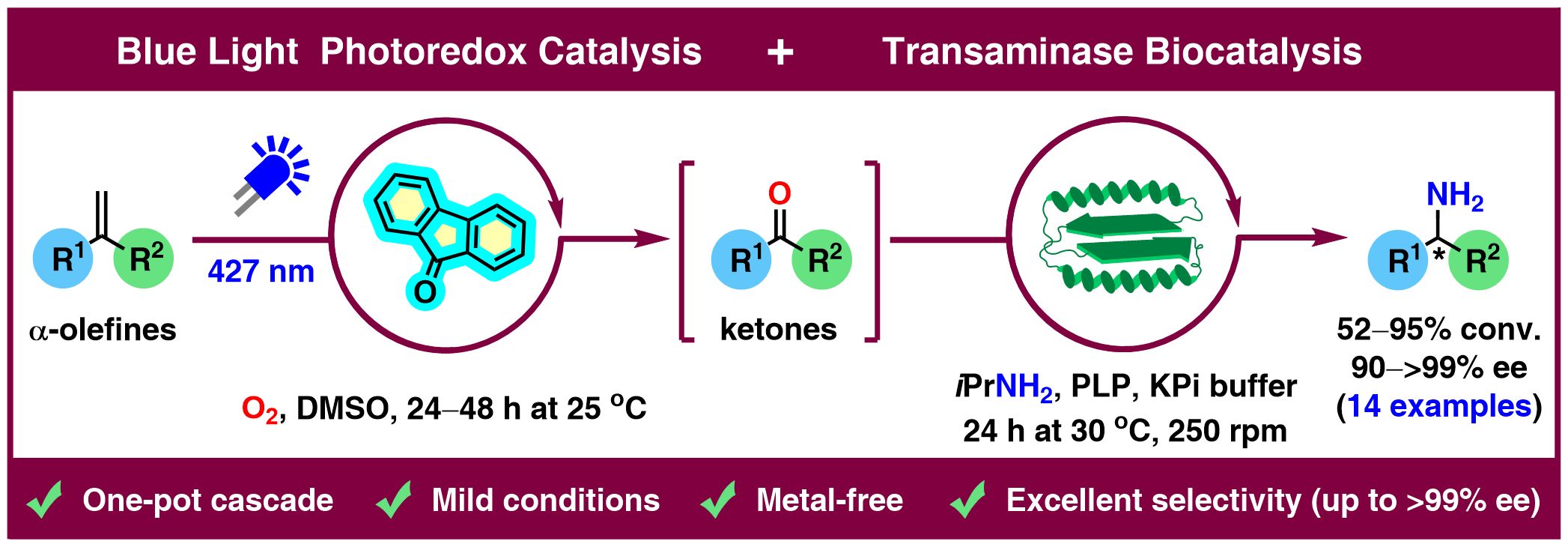
Merging photocatalysis with biocatalysis has recently emerged as a powerful strategy in chemical synthesis. Whereas photocatalysis enables the generation of reactive intermediates under mild conditions, biocatalysis provides exceptional selectivity with minimal byproducts. Herein, we report a one-pot/two-step photo-biocatalytic oxidation–reductive amination cascade that transforms α-methyl terminal alkenes into enantiomerically pure amines. In the initial step, 1,1-disubstituted (hetero)aryl alkyl olefins underwent oxidative cleavage to prochiral ketones using molecular oxygen under blue-light irradiation (427 nm) with 9-fluorenone as a transition-metal-free photocatalyst in dimethyl sulfoxide (DMSO), which simultaneously serves as solvent and quencher of in situ generated H2O2. Subsequent sequential combination with transaminase-catalyzed reductive amination furnished the targeted nonracemic amines in good-to-excellent overall conversions (52%–95%) and high-to-excellent enantiomeric excesses (90%–99.9%) in a stereocomplementary manner. The photo-biocatalytic cascade was successfully scaled up with α-methylstyrene (1.0 mmol), enabling the synthesis of the pharmaceutically relevant chiral precursor (R)-(+)-1-phenylethylamine in 93% conversion, 61% isolated yield, and >99% ee. Further derivatization of this key intermediate delivered the L-type calcium channel blocker fendiline in 77% isolated yield and 99% ee, without the need for costly transition metal catalysts, hazardous hydrogen gas, and high pressure.
doi: 10.1002/adsc.70219
2025
49. Synthetic strategies toward nefopam: a short review
M. Dąbrowski,* P. Borowiecki*
ARKIVOC 2025, 1, 202512420.
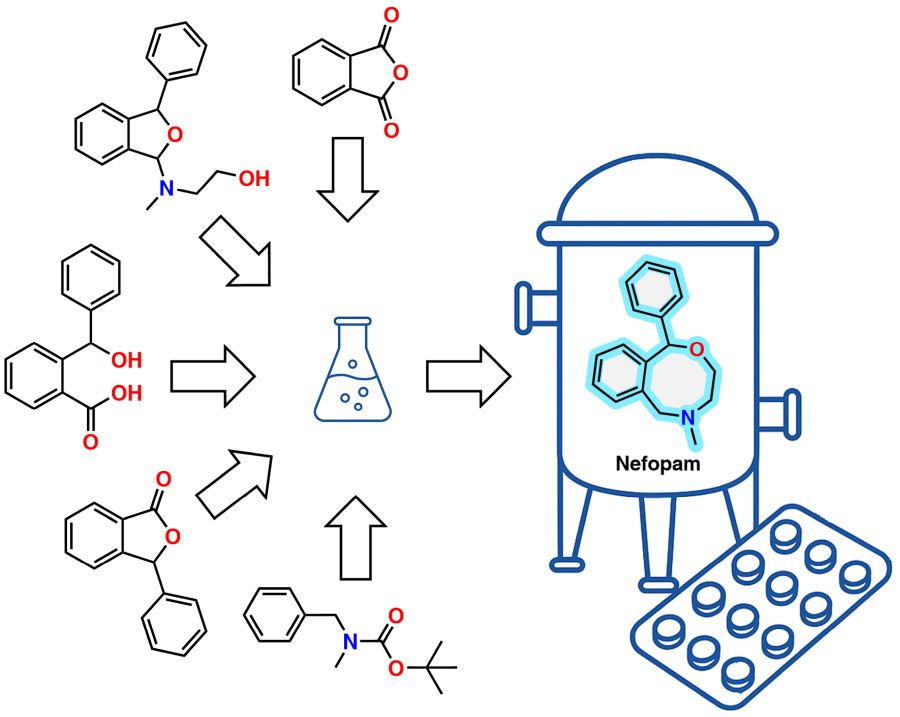
Nefopam is a centrally acting, non-opioid, and non-steroidal analgesic with both oral and parenteral bioavailability. Its clinical utility arises from a favorable side-effect profile relative to conventional analgesics, which has led to its adoption in the management of acute postoperative, dental, and renal pain, as well as for the prevention of postoperative shivering and treatment of intractable hiccups. Although nefopam has demonstrated efficacy in selected chronic pain conditions, such as migraine and facial pain, its therapeutic use remains predominantly restricted to acute pain settings. This short review provides a concise overview of synthetic strategies developed for nefopam and its structural analogues, with particular emphasis on their potential for adaptation to industrial-scale synthesis. The survey encompasses peer-reviewed research articles and patent documentations published between 1969 and 2025.
48. Development of Photo-Biocatalytic Redox System for Asymmetric Synthesis of Optically Active Amines Using Blue Light and Transaminases. A Case Study Toward Mavacamten
N. Antos, A. Rudzka, A. Hoser, T. Reiter, W. Kroutil, P. Borowiecki*
Adv. Synth. Catal. 2025, 367, e202500250.
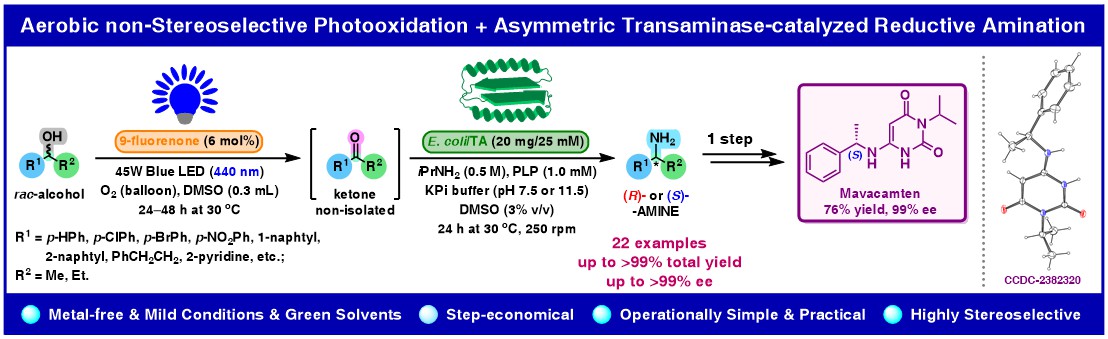
α-Chiral amines are versatile building blocks for the asymmetric synthesis of optically pure high-added-value chemicals, including pharmaceuticals, agrochemicals, and natural products. Herein, we report a one-pot, two-step sequential deracemization of racemic sec-alcohols to optically enriched primary amines throught a photo-biocatalytic oxidation-reductive amination linear cascade. In the first step, near-to-quantitative oxidation of a set of racemic (hetero)benzylic alcohols into prochiral ketones was accomplished using visible light photoredox catalysis relying on 440 nm blue LEDs irradiation, 9-fluorenone as a transition-metal-free photocatalyst, and O2-saturated dimethyl sulfoxide (DMSO) as the reaction medium and a quencher of the in situ generated hydrogen peroxide (H2O2). The intermediary ketones were further converted into the corresponding non-racemic amines with up to 99% conversion, high-to-excellent optical purity (90–99.9% ee), and complementary absolute configuration via a stereoselective reductive amination catalyzed by lyophilized E. coli cells containing the respective (R)- or (S)-selective recombinant transaminases (E. coli/TAs) in an aqueous KPi buffer in the presence of pyridoxal-5′-phosphate (PLP) as cofactor and isopropylamine (iPrNH2) as an amino group donor. The developed photo-biocatalytic system was scaled up for racemic 1-phenylethanol (1.0 mmol), furnishing (S)-(−)-1-phenylethylamine with 91% conv., 82% isolated yield and 99% ee. An exemplary functionalization of the obtained (S)-α-methylbenzylamine with 6-chloro-3-isopropylpyrimidine-2,4(1H,3H)-dione led to afford an enantiomerically enriched newly launched cardiac-specific myosin inhibitor mavacamten in 76% yield and 99% ee. In addition, a single-crystal X-ray diffraction (XRD) analysis was performed, furnishing the first crystal structure for the titled API.
47. Chemoenzymatic synthesis (Editorial)
P. Borowiecki* and S. Schmidt*
Commun. Chem. 2025, 8, 77.
Communications Chemistry is pleased to introduce a Collection of research works focused on recent developments within the interdisciplinary field of chemoenzymatic synthesis. Here, the Guest Editors highlight key themes and look towards the future of this research field.
46. Bienzymatic Dynamic Kinetic Resolution of Secondary Alcohols by Esterification/Racemization in Water
A. Rudzka, T. Reiter, W. Kroutil,* P. Borowiecki*
Angew. Chem. Int. Ed. 2025, 64, e202420133.
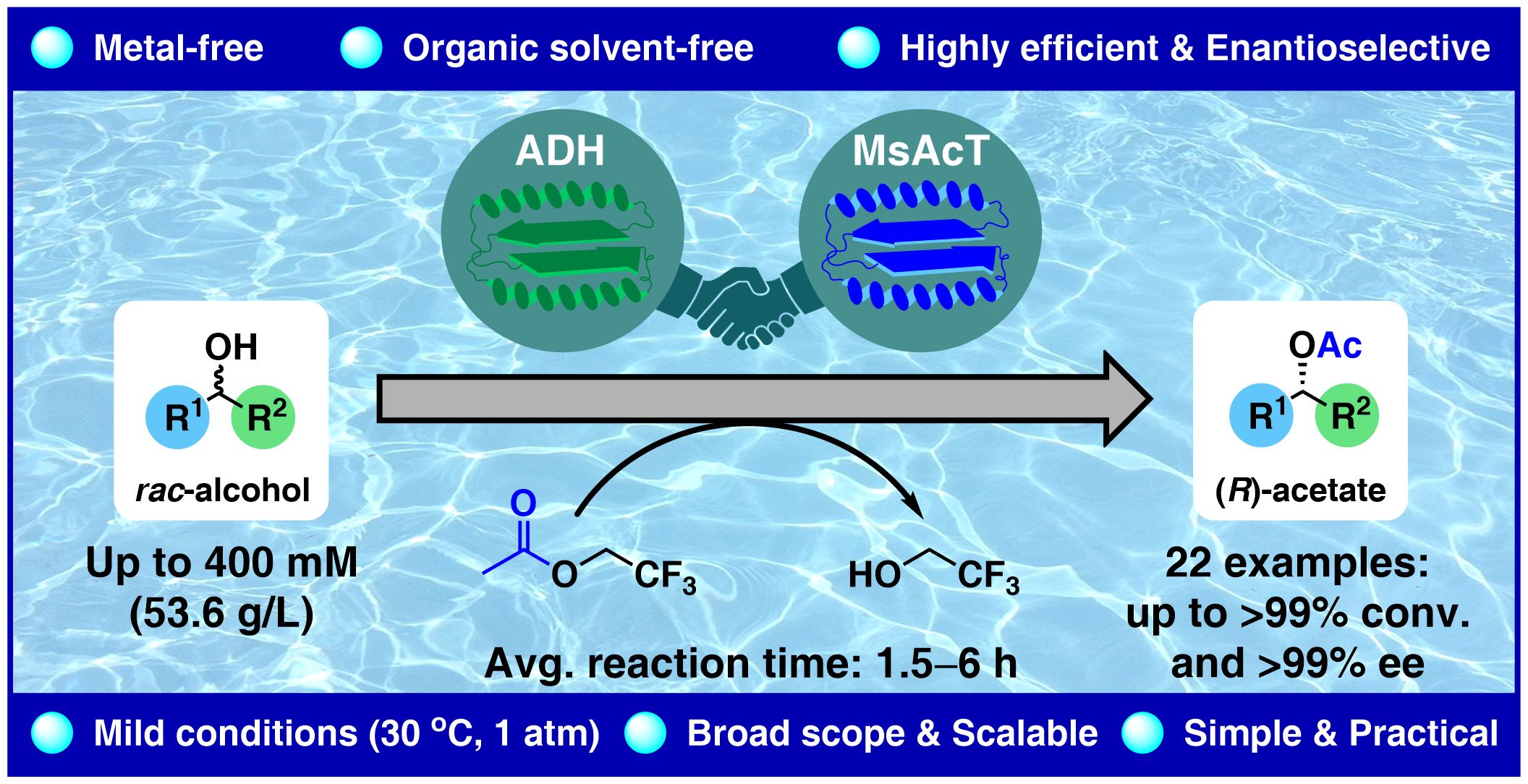
Dynamic kinetic resolution (DKR) is a key method used to prepare optically pure compounds in 100 % theoretical yield starting from racemic substrates by combining the interconversion of substrate enantiomers with an enantioselective transformation. Various chemoenzymatic DKR approaches have been developed to deracemize secondary alcohols, typically requiring an organic solvent to facilitate enantioselective acylation, primarily catalyzed by lipases, alongside racemization mediated by an achiral, non-enzymatic catalyst. Achieving both steps in an aqueous solution remained elusive. Herein, we report a DKR of racemic sec-alcohols in an aqueous solution requiring only two biocatalysts. The first key to success was to achieve fast racemization in a buffer employing a non-stereoselective variant of an alcohol dehydrogenase (Lk-ADH-Prince) via a hydrogen-borrowing oxidation-reduction sequence. Engineered variants of the acyltransferase from Mycobacterium smegmatis (MsAcT) enabled enantioselective acyl transfer in water. Besides the appropriate choice of the enzymes, identifying a suitable acyl donor was a second key to the success. The DKR was successfully demonstrated using (R)-selective MsAcT variants for a broad range of racemic (hetero)benzylic alcohols with 2,2,2-trifluoroethyl acetate as the acyl donor, yielding (R)-acetates with up to >99 % conv. and high-to-excellent optical purity (83–99.9 % ee). The (S)-acetates were accessible using a stereocomplementary (S)-selective MsAcT variant. Notably, substrate concentrations of up to 400 mM were tolerated in selected cases.
45. Introducing ... Paweł Borowiecki
P. Borowiecki*
Angew. Chem. Int. Ed. 2025, 64, e202424488.

“The most exciting thing about my research is exploring the remarkable catalytic potential of enzymes and witnessing the passion and motivation in the eyes of students and young co-workers, as they engage with scientific discovery… My first experiment was developing analog photographs in a darkroom I set up in the basement of my parents’ house…” Find out more about Paweł Borowiecki in his Introducing… Profile.
2024
44. An experimental and computational investigation of the cyclopentene-containing peptide-derived compounds: focus on pseudo-cyclic motifs via intramolecular interactions
J. Bojarska,* M. Breza, P. Borowiecki, I. D. Madura, K. Kaczmarek, Z. M. Ziora,* W. M. Wolf
R. Soc. Open Sci. 2024, 11, 40962.
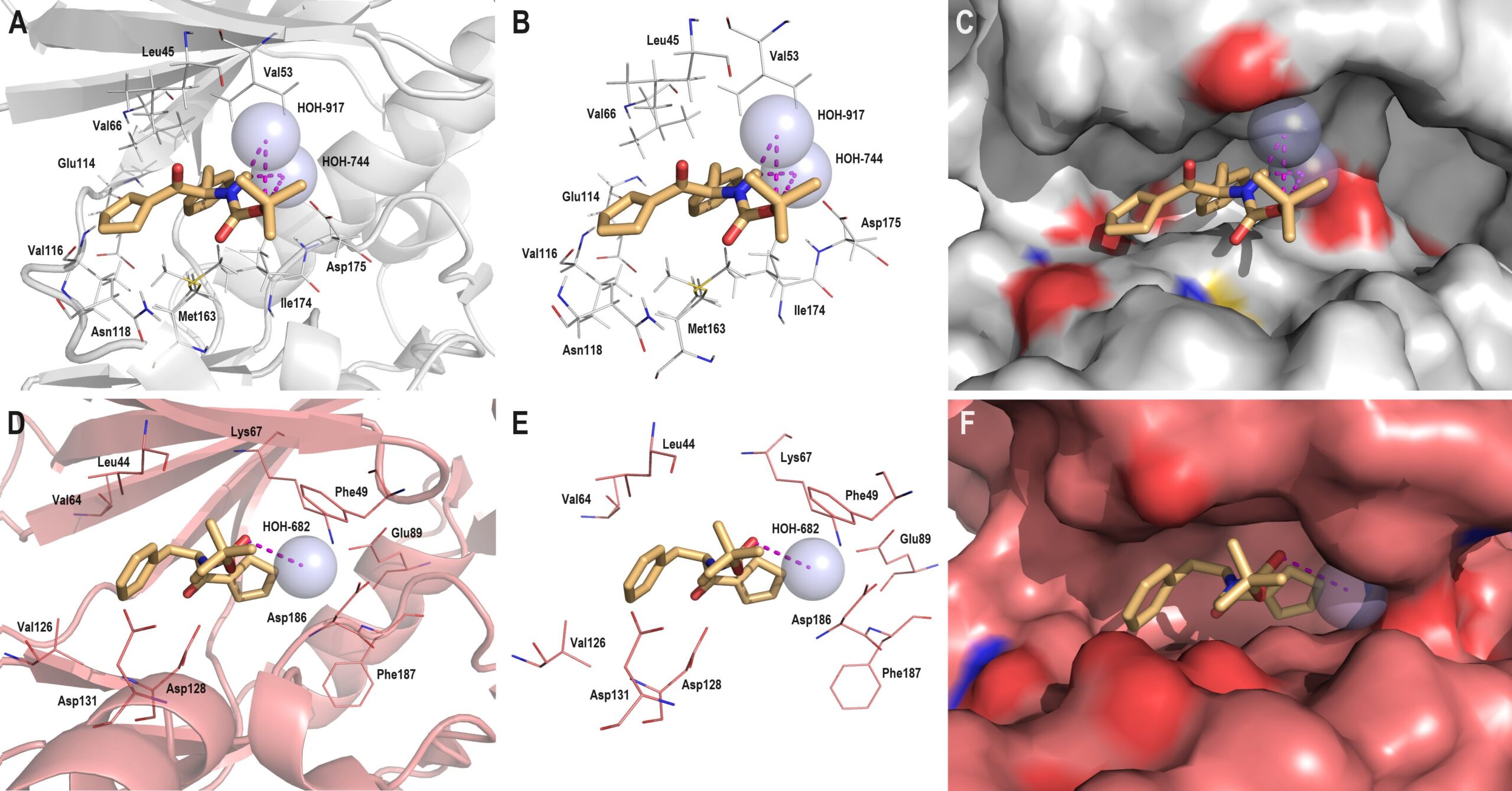
Conformational flexibility is one of the main disadvantages of peptide-based compounds. We focus on their molecular ‘chameleonicity’ related to forming pseudo-cyclic motifs via modulation of weak intramolecular interactions. It is an appealing strategy for controlling equilibrium between the polar open and the nonpolar closed conformations. Within this context, we report here the crystal structure of the (R)-(2-tert-butoxycarbonyl)amino-1-oxo-3-phenyl)propyl)-1-cyclopentene (1), synthesis of which in high yield was achieved by a facile multi-step protocol. Our Cambridge Structural Database (CSD) overview for the peptide-based crystals revealed the exclusivity of this compound from the viewpoint of the unusual pseudo-bicyclic system via C–H…O and C–O…π interactions, in which cyclopentene shields the amide bond. Notably, cyclopentene as a bioisostere of proline is an appealing scaffold in medicinal chemistry. An extensive combined experimental and computational study provided more profound insight into the supramolecular landscape of 1 with respect to similar derivatives deposited in the CSD, including the tendency of cyclopentene for the generation of pseudo-cyclic motifs through weak H-bonding and π-based intramolecular interactions. These weak interactions have been examined by either the quantum theory of ‘atoms-in-molecules’ (QTAIM) or complex Hirshfeld surface methodology, including enrichment ratios, molecular electrostatic potential surfaces and energy frameworks. In all analysed crystals, all types of H-bonded motifs involving cyclopentene are formed at all levels of supramolecular architecture. A library of cyclopentene-based H-bonding synthons is provided. A molecular docking study depicted vital interactions of cyclopentene with key amino acid residues inside the active sites of two prominent protein kinases, uncovering the therapeutic potential of 1 against breast cancer. To a large extent, dispersion forces have significance in stabilizing the supramolecular structure of both ligand and bio-complex ligand–protein. Finally, the satisfactory in silico bio-pharmacokinetic profile of 1 related to drug-likeness and blood–brain barrier permeation was also revealed.
doi: 10.1098/rsos.240962
43. Vinyl 3-(Dimethylamino)propanoate as an Irreversible Acyl Donor Reagent in a Chromatography-free Lipase-Catalyzed Kinetic Resolution of sec-Alcohols
B. Zdun, P. Borowiecki*
ChemBioChem 2024, 25, e202400394.
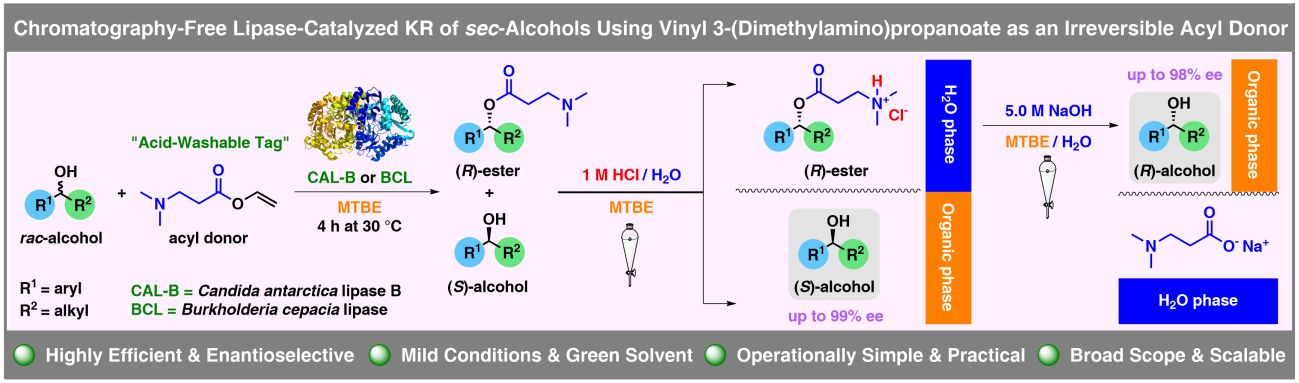
The reported chemoenzymatic strategy involves the employment of vinyl 3-(dimethylamino)propanoate as an irreversible acyl donor in a chromatography-free lipase-catalyzed kinetic resolution (KR) of racemic sec-alcohols. This biotransformation is achieved in a sequential manner using CAL-B to affect the kinetic resolution, followed by a simple acidic extractive work-up furnishing both KR products with excellent enantioselectivity (E>200; up to 98 % ee). The elaborated method eliminates a single-use silica gel chromatographic separation and significantly reduces organic solvent consumption to foster a more environmentally friendly chemical industry.
42. ADH-Catalyzed Biooxidation of (Hetero)aromatic sec-Alcohols to Ketones Employing Vinyl Acetate as Acetaldehyde Surrogate
A. Rudzka, T. Reiter, W. Kroutil, P. Borowiecki*
ChemCatChem 2024, 16, e202400803.
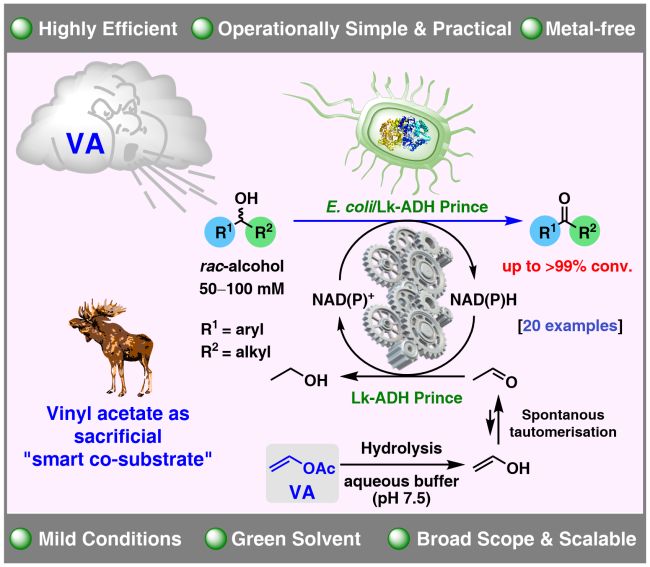
The oxidation of sec-alcohols is a common reaction in organic chemistry. We report a biocatalytic approach for oxidizing racemic (hetero)aromatic sec-alcohols to the corresponding ketones. The reaction relies on employing freeze-dried E. coli cells containing a recombinant variant of an alcohol dehydrogenase deduced from Lactobacillus kefir (E. coli/Lk-ADH Prince) and vinyl acetate as an in situ acetaldehyde surrogate as oxidant. Biooxidations of a set of 20 racemic (hetero)aromatic alcohols were carried out in the presence of a catalytic amount of NADP+ cofactor, the biocatalyst, and vinyl acetate in an aqueous medium to generate the corresponding ketones with up to >99 % conv. Preparative scale (1.0 mmol; 100 mM in 10 mL) reactions led to obtaining ketones in the 56–83 % isolated yield range.
41. One-Pot Sequential Two-Step Photo-Biocatalytic Deracemization of sec-Alcohols Combining Photocatalytic Oxidation and Bioreduction
A. Rudzka, N. Antos, T. Reiter, W. Kroutil, P. Borowiecki*
ACS Catal. 2024, 14, 1808–1823.
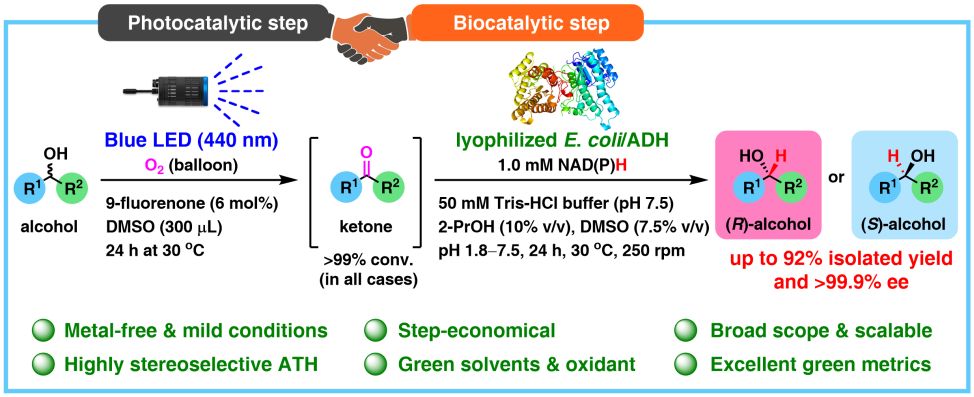
Chiral alcohols are versatile building blocks and are of particular interest in the asymmetric synthesis of nonracemic active pharmaceutical ingredients, agrochemicals, fragrances, flavors, natural products, etc. Herein, we report on a “one-pot sequential two-step” concurrent oxidation–reduction photobiocatalytic process to synthesize enantiomerically enriched alcohols. In this regard, an efficient photocatalytic system based on irradiation with 440 nm blue LEDs in the presence of 9-fluorenone as a metal-free photocatalyst and molecular oxygen as the terminal oxidant in dry DMSO as the hydrogen peroxide-neutralizing agent was used to oxidize a broad range of racemic (hetero)benzylic alcohols into prochiral ketones quantitively (>99% conv.). The in situ formed carbonyl compounds were subsequently converted into the corresponding chiral alcohols via a sequential biocatalytic transhydrogenation catalyzed by lyophilized E. coli cells overexpressing highly stereoselective and stereocomplementary recombinant alcohol dehydrogenases (ADHs) originated from Rhodococcus ruber (E. coli/ADH-A) or Rhodococcus erythropolis (E. coli/ReADH) to obtain (S)-alcohols and Lactobacillus kefir (E. coli/Lk-ADH) or KRED-110 to obtain (R)-alcohols, respectively. Overall, the elaborated photobiocatalytic deracemization of racemic alcohols using a 9-fluorenone-O2-blue LED-DMSO-E. coli/ADH system carried out on a semipreparative scale (0.25 mmol; 63 mM final conc. in 4 mL) at room temperature yielded nonracemic aryl alcohols with 82–99.9% conv., in up to 92% isolated yield, with 97–99.9% ee and complementary chirality.
2023
40. Biocatalytic characterization of an alcohol dehydrogenase variant deduced from Lactobacillus kefir in asymmetric hydrogen transfer
A. Rudzka, B. Zdun, N. Antos, L. Martínez Montero, T. Reiter, W. Kroutil, P. Borowiecki*
Commun. Chem. 2023, 6, 1−14.
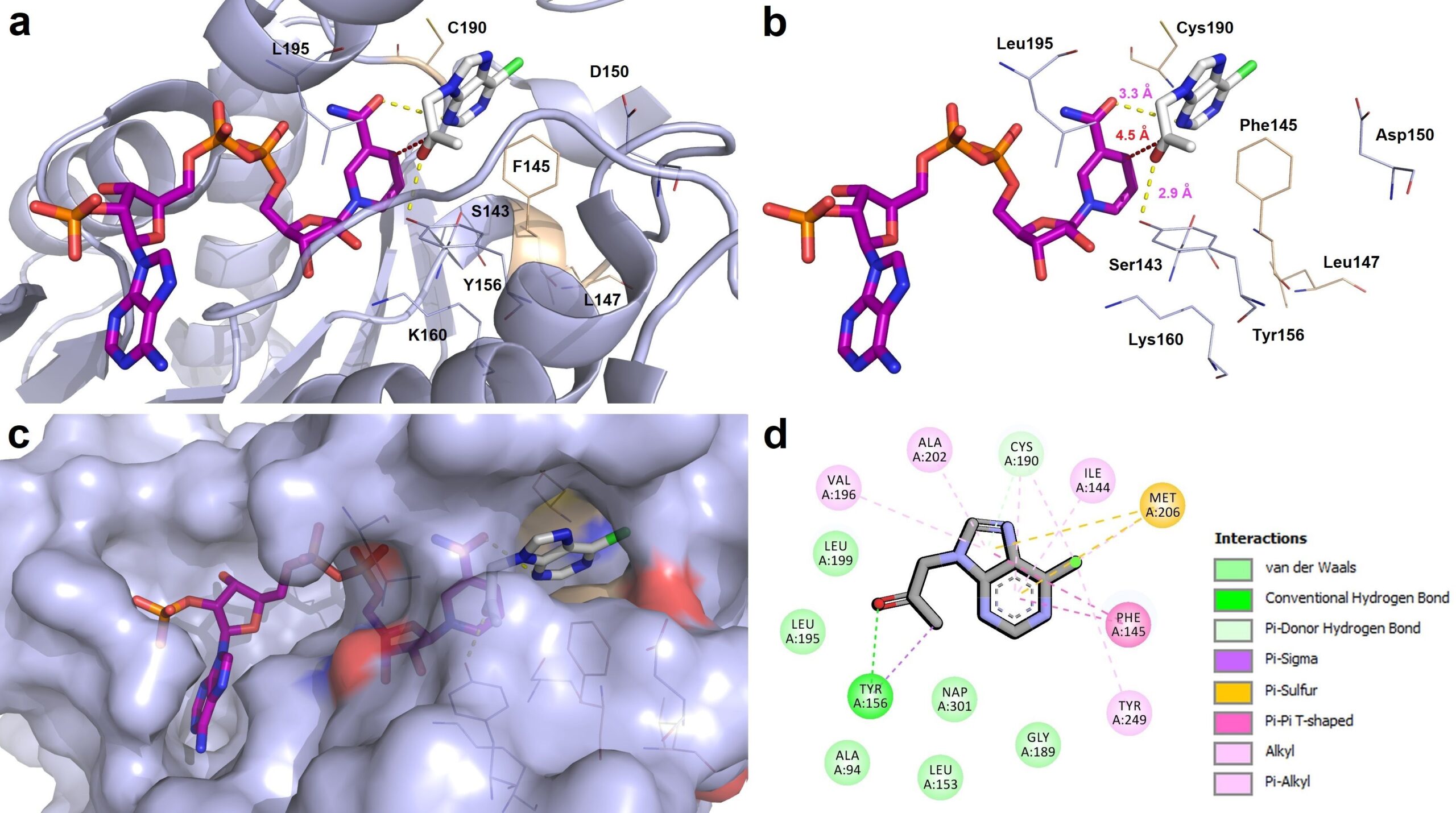
Hydrogen transfer biocatalysts to prepare optically pure alcohols are in need, especially when it comes to sterically demanding ketones, whereof the bioreduced products are either essential precursors of pharmaceutically relevant compounds or constitute APIs themselves. In this study, we report on the biocatalytic potential of an anti-Prelog (R)-specific Lactobacillus kefir ADH variant (Lk-ADH-E145F-F147L-Y190C, named Lk-ADH Prince) employed as E. coli/ADH whole-cell biocatalyst and its characterization for stereoselective reduction of prochiral carbonyl substrates. Key enzymatic reaction parameters, including the reaction medium, evaluation of cofactor-dependency, organic co-solvent tolerance, and substrate loading, were determined employing the drug pentoxifylline as a model prochiral ketone. Furthermore, to tap the substrate scope of Lk-ADH Prince in hydrogen transfer reactions, a broad range of 34 carbonylic derivatives was screened. Our data demonstrate that E. coli/Lk-ADH Prince exhibits activity toward a variety of structurally different ketones, furnishing optically active alcohol products at the high conversion of 65–99.9% and in moderate-to-high isolated yields (38–91%) with excellent anti-Prelog (R)-stereoselectivity (up to >99% ee) at substrate concentrations up to 100 mM.
39. Chemoenzymatic synthesis of tenofovir
B. Zdun, T. Reiter, W. Kroutil, P. Borowiecki*
J. Org. Chem. 2023, 88, 11045−11055.
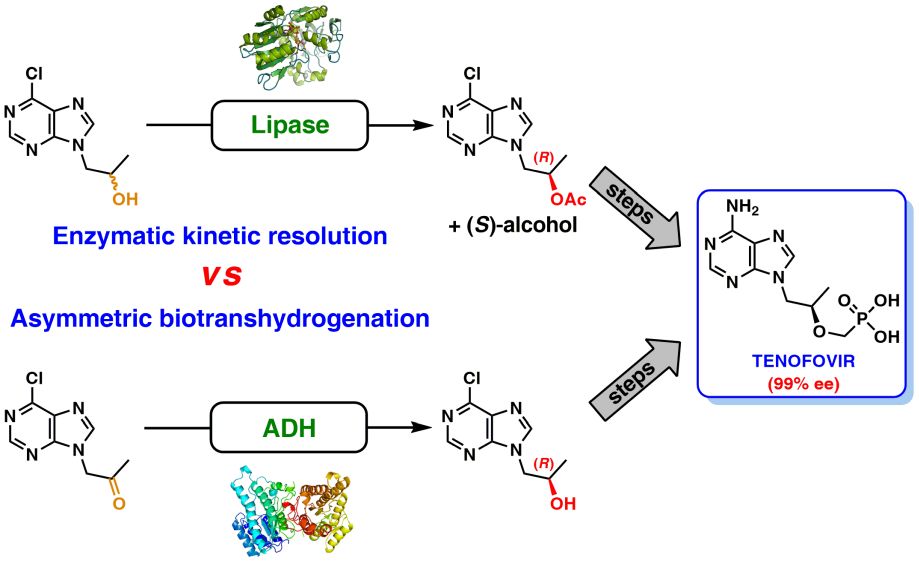
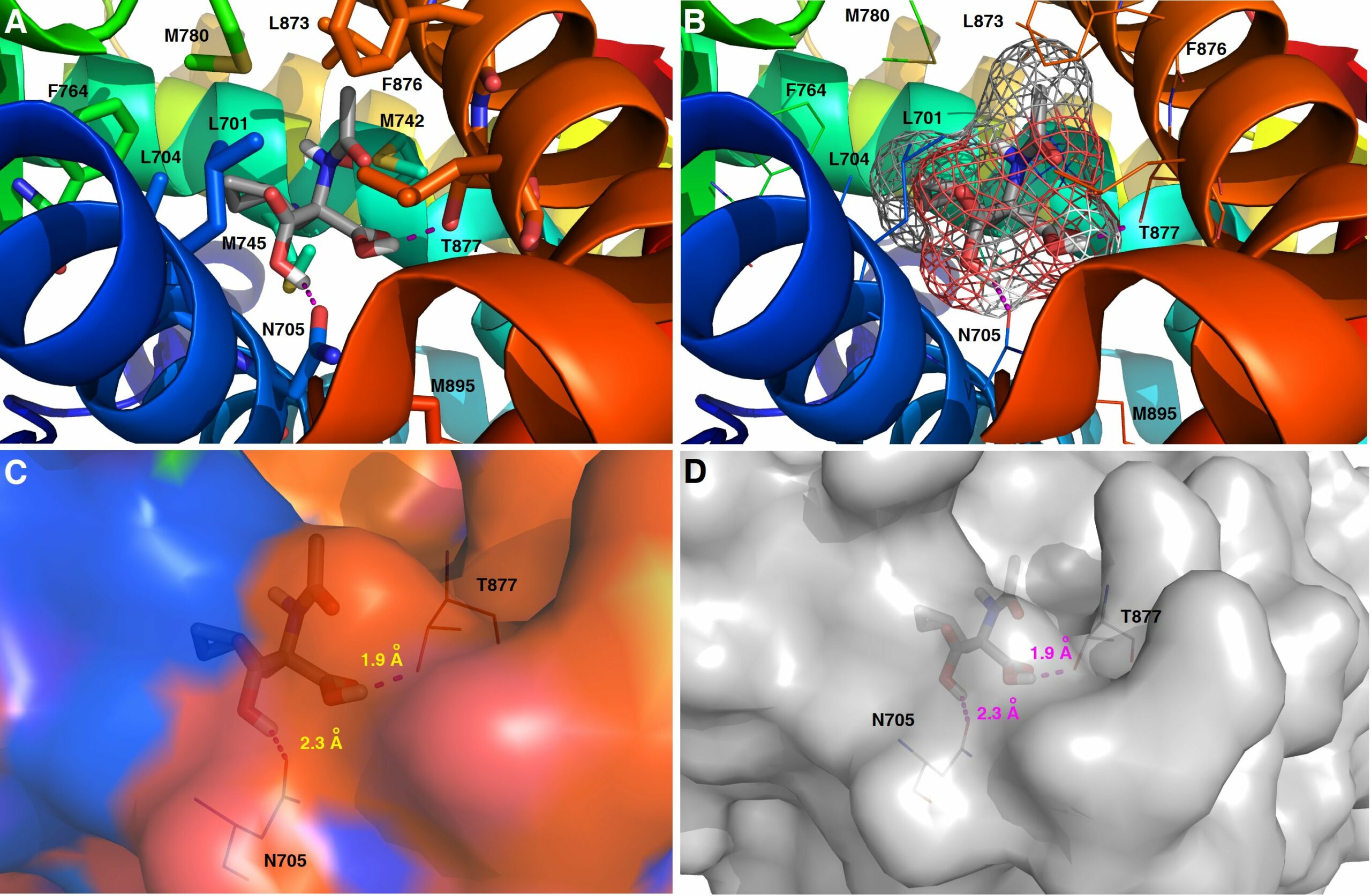
We report on novel chemoenzymatic routes toward tenofovir using low-cost starting materials and commercial or homemade enzyme preparations as biocatalysts. The biocatalytic key step was accomplished either via stereoselective reduction using an alcohol dehydrogenase or via kinetic resolution using a lipase. By employing a suspension of immobilized lipase from Burkholderia cepacia (Amano PS-IM) in a mixture of vinyl acetate and toluene, the desired (R)-ester (99% ee) was obtained on a 500 mg scale (60 mM) in 47% yield. Alternatively, stereoselective reduction of 1-(6-chloro-9H-purin-9-yl) propan-2-one (84 mg, 100 mM) catalyzed by lyophilized E. coli cells harboring recombinant alcohol dehydrogenase (ADH) from Lactobacillus kefir (E. coli/Lk-ADH Prince) allowed one to reach quantitative conversion, 86% yield and excellent optical purity (>99% ee) of the corresponding (R)-alcohol. The key (R)-intermediate was transformed into tenofovir through “one-pot” aminolysis–hydrolysis of (R)-acetate in NH3-saturated methanol, alkylation of the resulting (R)-alcohol with tosylated diethyl(hydroxymethyl) phosphonate, and bromotrimethylsilane (TMSBr)-mediated cleavage of the formed phosphonate ester into the free phosphonic acid. The elaborated enzymatic strategy could be applicable in the asymmetric synthesis of tenofovir prodrug derivatives, including 5′-disoproxil fumarate (TDF, Viread) and 5′-alafenamide (TAF, Vemlidy). The molecular basis of the stereoselectivity of the employed ADHs was revealed by molecular docking studies.
38. Synthesis and Anticancer Activity of Novel Dual Inhibitors of Human Protein Kinases CK2 and PIM-1
P. Wińska,* M. Wielechowska, M. Koronkiewicz, P. Borowiecki,*
Pharmaceutics 2023, 15, 199.
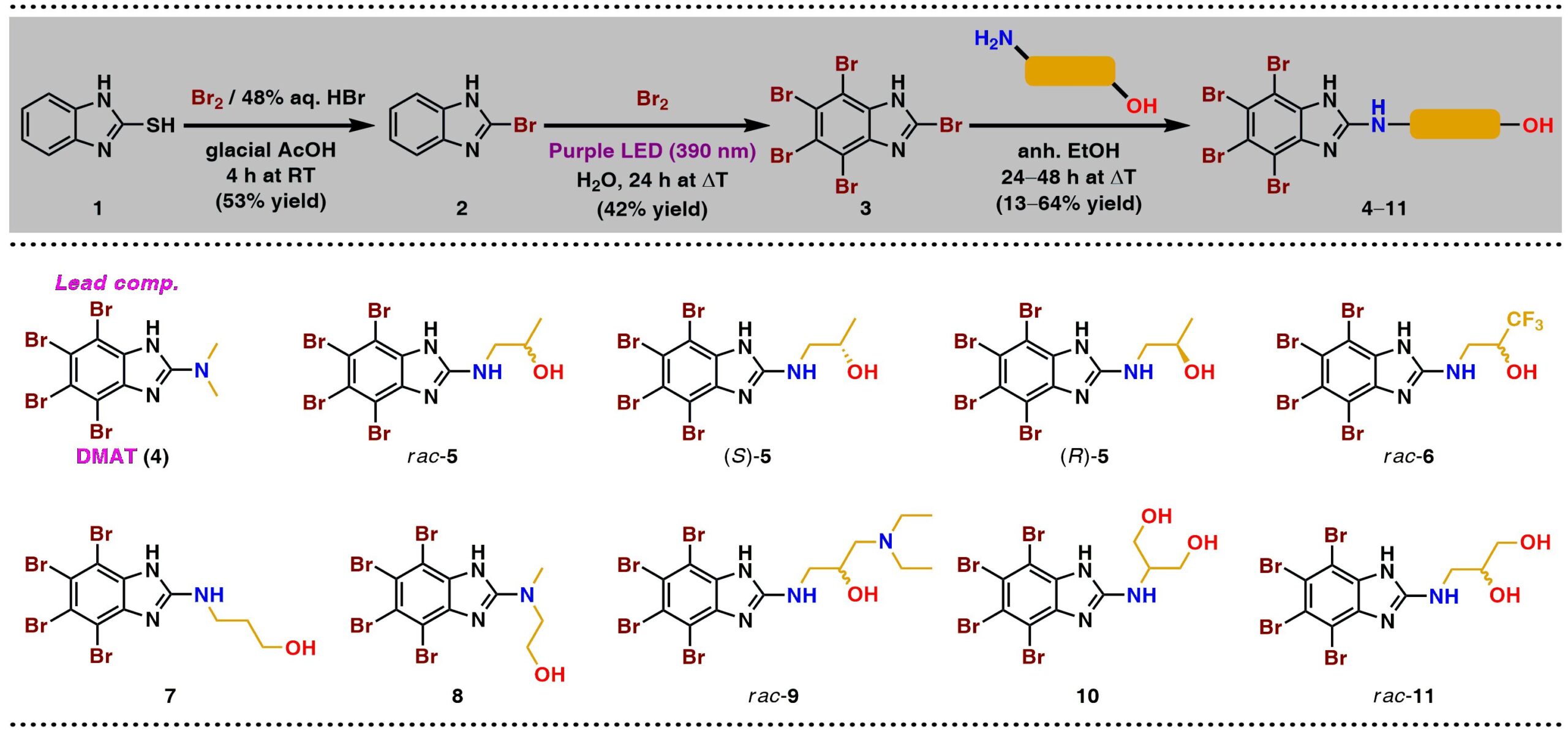
CK2 and PIM-1 are serine/threonine kinases involved in the regulation of many essential processes, such as proliferation, differentiation, and apoptosis. Inhibition of CK2 and PIM-1 kinase activity has been shown to significantly reduce the viability of cancer cells by inducing apoptosis. A series of novel amino alcohol derivatives of parental DMAT were designed and synthesized as potent dual CK2/PIM-1 inhibitors. Concomitantly with the inhibition studies toward recombinant CK2 and PIM-1, the influence of the obtained compounds on the viability of three human carcinoma cell lines, i.e., acute lymphoblastic leukemia (CCRF-CEM), human chronic myelogenous leukemia (K-562), and breast cancer (MCF-7), as well as non-cancerous cells (Vero), was evaluated using an MTT assay. Induction of apoptosis and cell cycle progression after treatment with the most active compound and a lead compound were studied by flow-cytometry-based assay. Additionally, autophagy induction in K-562 cells and intracellular inhibition of CK2 and PIM-1 in all the tested cell lines were evaluated by qualitative/quantitative fluorescence-based assay and Western blot method, respectively. Among the newly developed inhibitors, 1,1,1-trifluoro-3-[(4,5,6,7-tetrabromo-1H-benzimidazol-2-yl)amino]propan-2-ol demonstrates the highest selectivity and the most prominent proapoptotic properties towards the studied cancer cells, especially towards acute lymphoblastic leukemia, in addition to inducing autophagy in K-562 cells.
37. Effective monitoring of Platinum-DNA adducts formation under simulated physiological conditions by CE-ICP-MS/MS
J. Zajda,* P. Borowiecki, M. Matczuk
Talanta 2023, 264, 124749.
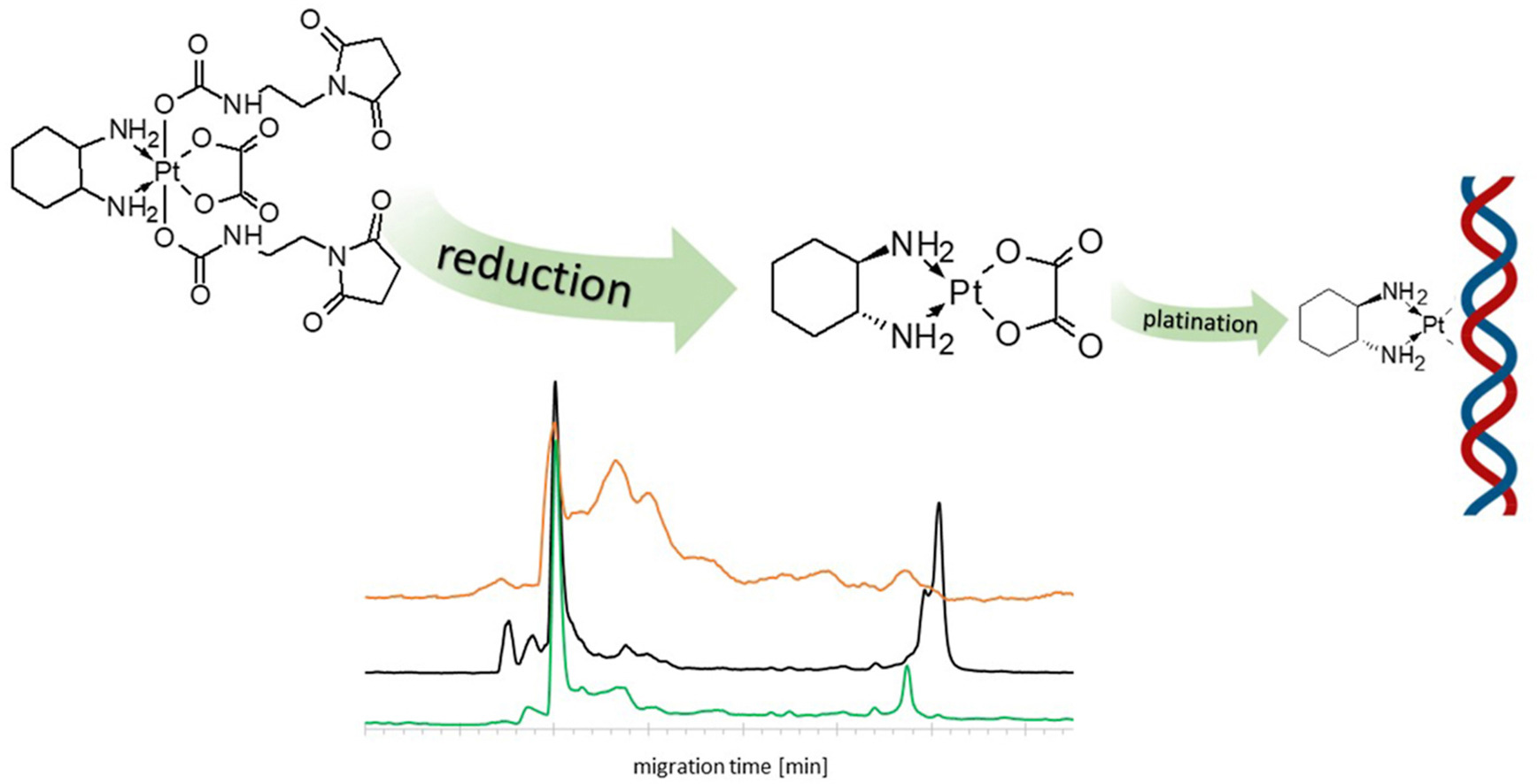
The leading Pt(II)-based anitcancer drugs have been used for decades; however, chemotherapy with their application is burdened with severe side effects. The administration of compounds capable of DNA platination in the form of prodrugs has the potential to overcome the drawbacks associated with their use. Progress toward their clinical application depends on establishing proper methodologies that would allow assessing their ability to bind to DNA in the biological environment. Herein, we propose implementing the approach based on the hyphenation of capillary electrophoresis with inductively coupled plasma tandem mass spectrometry (CE-ICP-MS/MS) for studying Pt-DNA adduct formation. The presented methodology opens the possibility to employ the multielement monitoring for studying the differences in the behavior of Pt(II) and Pt(IV) complexes and, interestingly, revealed the formation of various adducts with DNA and cytosol components for the latter one.
2022
36. Chemoenzymatic Synthesis of Optically Active Alcohols Possessing 1,2,3,4-Tetrahydroquinoline Moiety Employing Lipases or Variants of the Acyltransferase from Mycobacterium smegmatis
B. Zdun, I. Kopińska, M. Dranka, T. Reiter, W. Kroutil, P. Borowiecki*
Catalysts 2022, 12, 1610.
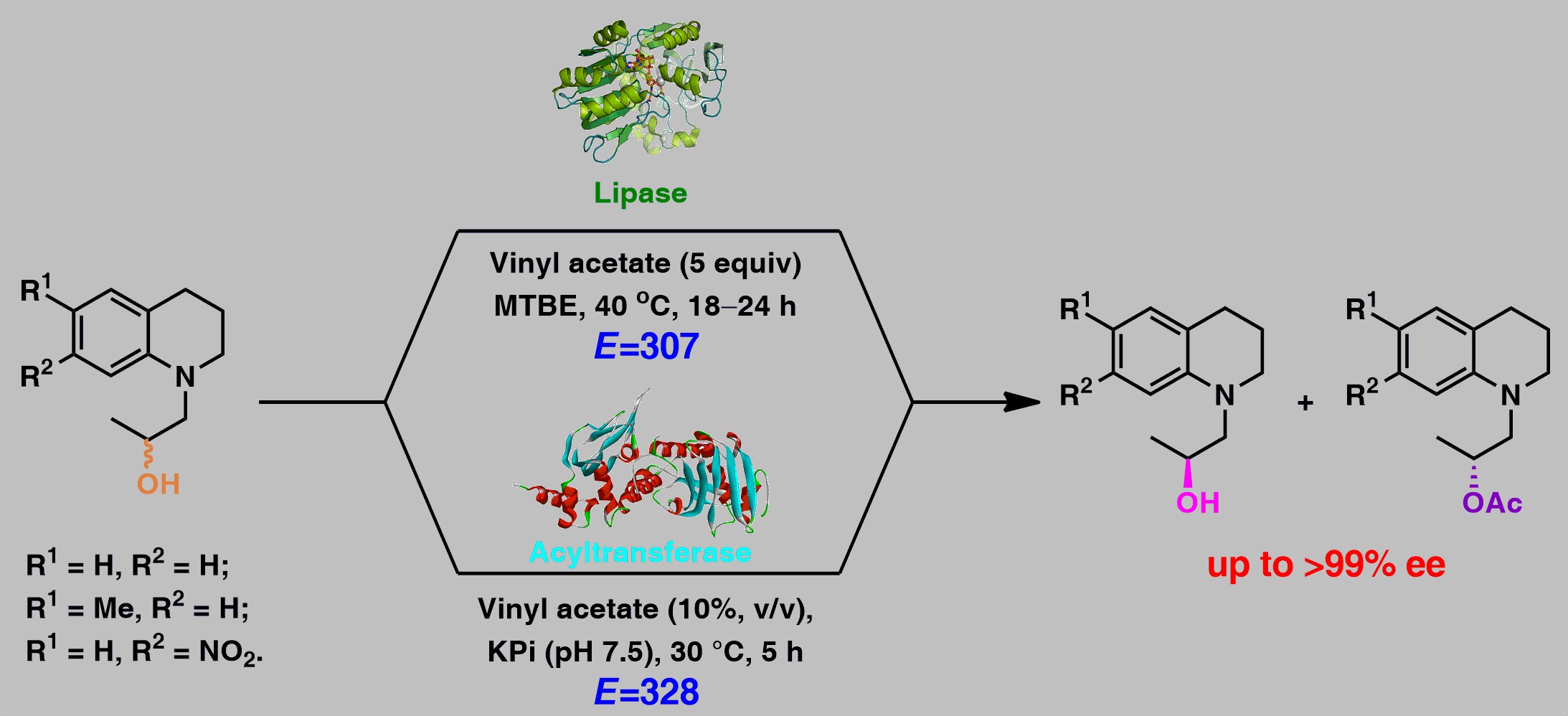
The enzymatic kinetic resolution (EKR) of racemic alcohols or esters is a broadly recognized methodology for the preparation of these compounds in optically active form. Although EKR approaches have been developed for the enantioselective transesterification of a vast number of secondary alcohols or hydrolysis of their respective esters, to date, there is no report of bio- or chemo-catalytic asymmetric synthesis of non-racemic alcohols possessing 1,2,3,4-tetrahydroquinoline moiety, which are valuable building blocks for the pharmaceutical industry. In this work, the kinetic resolution of a set of racemic 1,2,3,4-tetrahydroquinoline-propan-2-ols was successfully carried out in neat organic solvents (in the case of CAL-B and BCL) or in water (in the case of MsAcT single variants) using immobilized lipases from Candida antarctica type B (CAL-B) and Burkholderia cepacia (BCL) or engineered acyltransferase variants from Mycobacterium smegmatis (MsAcT) as the biocatalysts and vinyl acetate as irreversible acyl donor, yielding enantiomerically enriched (S)-alcohols and the corresponding (R)-acetates with E-values up to 328 and excellent optical purities (>99% ee). In general, higher ee-values were observed in the reactions catalyzed by lipases; however, the rates of the reactions were significantly better in the case of MsAcT-catalyzed enantioselective transesterifications. Interestingly, we have experimentally proved that enantiomerically enriched 1-(7-nitro-3,4-dihydroquinolin-1(2H)-yl)propan-2-ol undergoes spontaneous amplification of optical purity under achiral chromatographic conditions.
35. Supramolecular synthon hierarchy in cyclopropyl-containing peptide-derived compounds
Bojarska, M. Breza, M. Remko, P. Borowiecki, A. Fruziński, I. D. Madura, K. Kaczmarek, Z. Leśnikowski, A. Kraj, P. Zielenkiewicz, W. M. Wolf
CrystEngComm 2022, 24, 8372-8389.
Considering the increasing importance of cyclopropyl-containing peptide-derived compounds in the pharmaceutical industry, herein, we report the crystal engineering of a series of novel derivatives, i.e., diethyl 2-acetamido-2-(cyclopropylmethyl)malonate (1), 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid (2) [Ac-b-cyclopropyl-(R,S)-Ala-OH], 2-(cyclopropylmethyl)-2-acetamidopropanedioic acid hydrate (3), 2-acetamido-3-cyclopropylpropanoic acid (4), and (2S)-2-[cyclopropyl(9H-luoren-9-ylmethoxycarbonyl)amino]propanoic acid (5) [Fmoc-b-cyclopropyl-(S)-Ala-OH]. Although several cyclopropyl-based peptide-derived structures have been reported in the literature, to the best of our knowledge, studies on the synthon hierarchy in this class of structures are limited. Thus, to address this gap, herein, we report a multidisciplinary study to shed light on the role of cyclopropyl synthons in (bio)supramolecular assemblies, opening a new vista for the further applications of this unique scaffold. The synthesis was achieved via a multi-step protocol in good yield and the structures were determined by single-crystal X-ray diffraction. The diverse supramolecular synthons responsible for the arrangement of molecules in the crystal lattice of either new compounds or all cyclopropyl-containing peptide-derived solids deposited in the CSD thus far were specified and summarized, building a library. The self-assembly is directed by the cooperative interplay of H-bonds and π-stacking interactions. Quantum-chemical computational studies revealed that the cyclopropyl ring is capable of π(C=O)⋯σ(cyclopropyl), π (arom)⋯σ*(cyclopropyl), π*(arom)⋯σ*(cyclopropyl) and π*(arom)⋯σ(cyclopropyl) interactions. The molecular docking study delineated the C–H⋯C(cyclopropyl) and C–H(cyclopropyl)⋯π interactions of the cyclopropyl moiety with the essential amino acid residues inside the active pocket of the human androgen receptor, highlighting the vital role of cyclopropyl in the supramolecular landscape of the bio-complex. Indeed, 2 shows a significant docking score with effective binding affinity, and thus is a promising candidate for prostate cancer prevention or management.
doi: 10.1039/D2CE01231F
34. Chemoenzymatic Synthesis of Optically Active Ethereal Analog of iso-Moramide—A Novel Potentially Powerful Analgesic
P. Borowiecki,*
Int. J. Mol. Sci. 2022, 23, 11803.
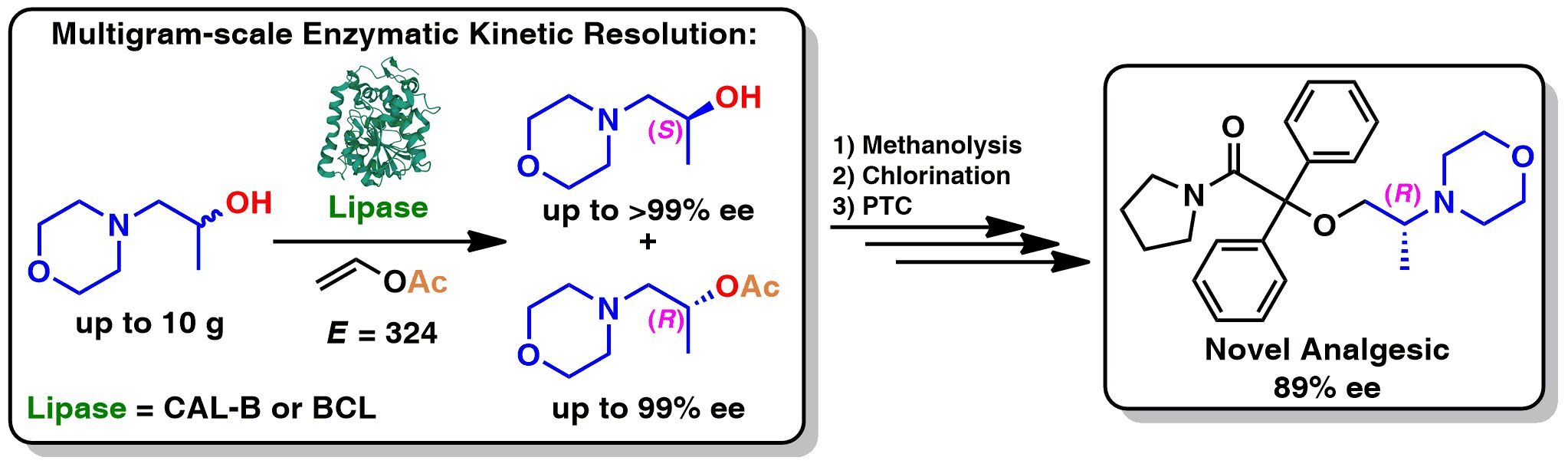
To develop potent and safer analgesics, we designed and synthesized a novel enantiomerically enriched ethereal analog of (R)-iso-moramide, namely 2-[(2R)-2-(morpholin-4-yl)propoxy]-2,2-diphenyl-1-(pyrrolidin-1-yl)ethan-1-one. The titled active agent can potentially serve as a powerful synthetic opiate with an improved affinity and selectivity toward opioid receptors (ORs). This hypothesis was postulated based on docking studies regarding the respective complexes between the designed ligand and mu-OR, delta-OR, and kappa-OR. The key step of the elaborated asymmetric synthesis of novel analog involves lipase-catalyzed kinetic resolution of racemic 1-(morpholin-4-yl)propan-2-ol, which was accomplished on a 10 g scale via an enantioselective transesterification employing vinyl acetate as an irreversible acyl donor in tert-butyl methyl ether (MTBE) as the co-solvent. Next, the obtained homochiral (S)-(+)-morpholino-alcohol (>99% ee) was functionalized into corresponding chloro-derivative using thionyl chloride (SOCl2) or the Appel reaction conditions. Further transformation with N-diphenylacetyl-1-pyrrolidine under phase-transfer catalysis (PTC) conditions using O2-saturated DMSO/NaOH mixture as an oxidant furnished the desired levorotatory isomer of the title product isolated in 26% total yield after three steps, and with 89% ee. The absolute configuration of the key-intermediate of (R)-(–)-iso-moramide was determined using a modified form of Mosher’s methodology. The preparation of the optically active dextrorotatory isomer of the titled product (87% ee) was carried out essentially by the same route, utilizing (R)-(–)-1-(morpholin-4-yl)propan-2-ol (98% ee) as a key intermediate. The spectroscopic characterization of the ethereal analog of iso-moramide and the enantioselective retention relationship of its enantiomers using HPLC on the cellulose-based chiral stationary phase were performed. Moreover, as a proof-of-principle, single-crystal X-ray diffraction (XRD) analysis of the synthesized 2-[(2R)-2-(morpholin-4-yl)propoxy]-2,2-diphenyl-1-(pyrrolidin-1-yl)ethan-1-one is reported.
33. Development of a novel chemoenzymatic route to enantiomerically enriched β-adrenolytic agents. A case study toward propranolol, alprenolol, pindolol, carazolol, moprolol, and metoprolol
P. Borowiecki,* B. Zdun, N. Popow, M. Wiklińska, T. Reiter, W. Kroutil
RSC Adv. 2022, 12, 22150–22160.
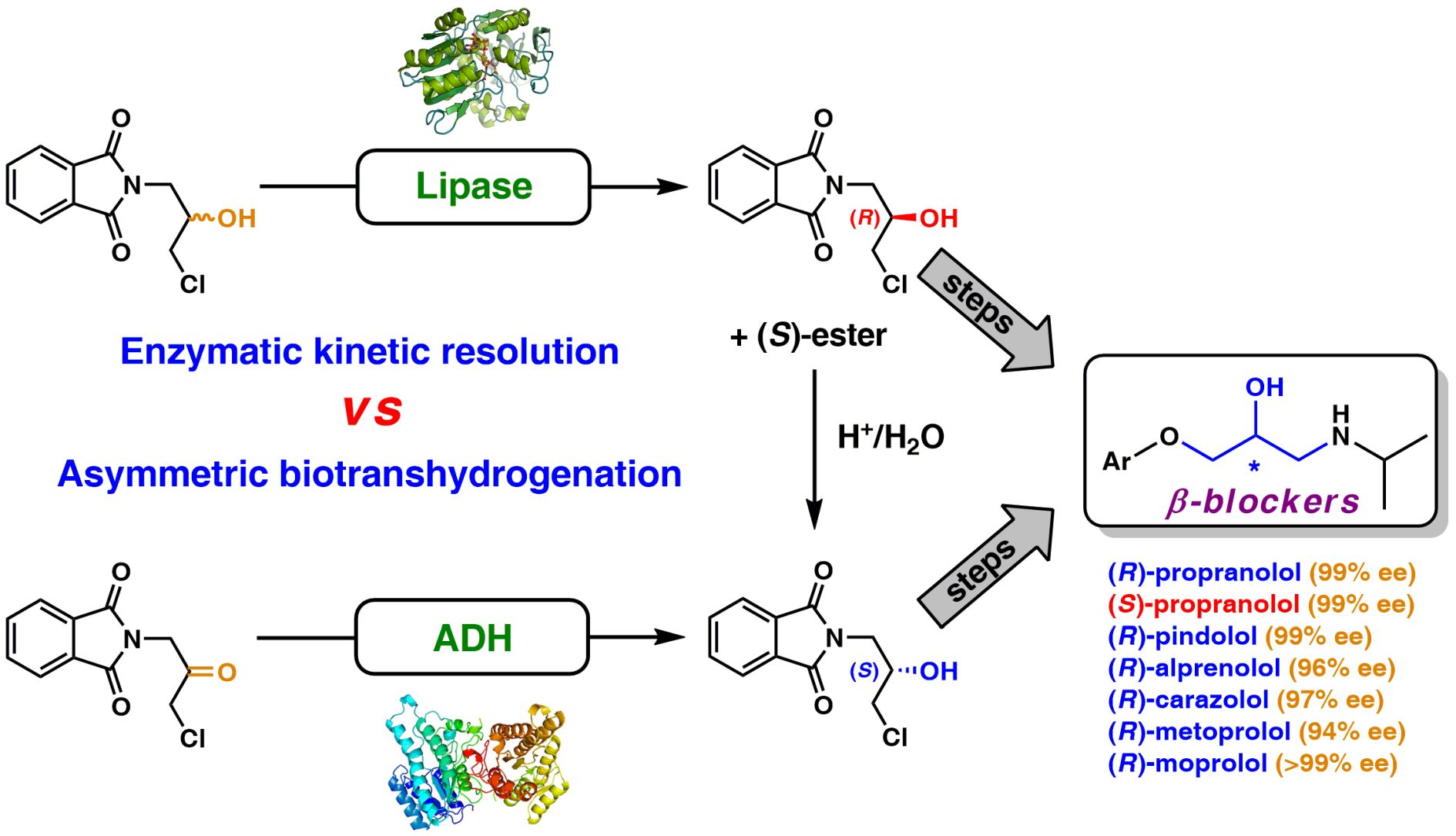
Efficient chemoenzymatic routes toward the synthesis of both enantiomers of adrenergic β-blockers were accomplished by identifying a central chiral building block, which was first prepared using lipase-catalyzed kinetic resolution (KR, Amano PS-IM) as the asymmetric step at a five gram-scale (209 mM conc.). The enantiopure (R)-chlorohydrin (>99% ee) subsequently obtained was used for the synthesis of a series of model (R)-(+)-β-blockers (i.e., propranolol, alprenolol, pindolol, carazolol, moprolol, and metoprolol), which were produced with enantiomeric excess in the range of 96–99.9%. The pharmaceutically relevant (S)-counterpart, taking propranolol as a model, was synthesized in excellent enantiomeric purity (99% ee) via acetolysis of the respective enantiomerically pure (R)-mesylate by using cesium acetate and a catalytic amount of 18-Crown-6, followed by acidic hydrolysis of the formed (S)-acetate. Alternatively, asymmetric reduction of a prochiral ketone, namely 2-(3-chloro-2-oxopropyl)-1H-isoindole-1,3(2H)-dione, was performed using lyophilized E. coli cells harboring overexpressed recombinant alcohol dehydrogenase from Lactobacillus kefir (E. coli/Lk-ADH-Lica) giving the corresponding chlorohydrin with >99% ee. Setting the stereocenter early in the synthesis and performing a 4-step reaction sequence in a ‘one-pot two-step’ procedure allowed the design of a ‘step-economic’ route with a potential dramatic improvement in process efficiency. The synthetic method can serve for the preparation of a broad scope of enantiomerically enriched β-blockers, the chemical structures of which rely on the common ahydroxy-N-isopropylamine moiety, and in this sense, might be industrially attractive.
doi: 10.1039/d2ra04302e
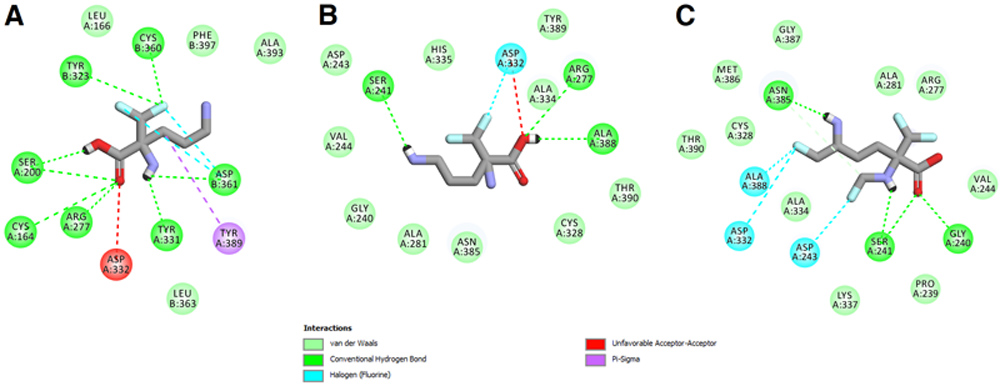
32. Biocatalytic Hydrogen-Transfer To Access Enantiomerically Pure Proxyphylline, Xanthinol, and Diprophylline
P. Borowiecki,* A. Rudzka, T. Reiter, W. Kroutil
Bioorg. Chem. 2022, 127, 105967–105978.
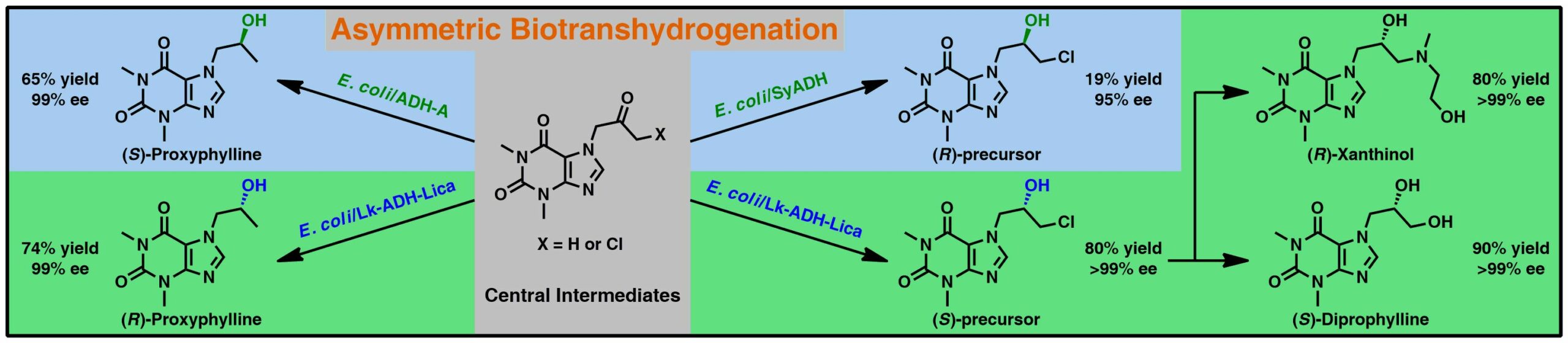
Alcohol dehydrogenases (ADHs; EC 1.1.1.1) have been widely used for the reversible redox reactions of carbonyl compounds (i.e., aldehydes and ketones) and primary or secondary alcohols, often resulting in optically pure hydroxyl products with high added value. In this work, we report a concise chemoenzymatic route toward xanthine-based enantiomerically pure active pharmaceutical ingredients (API) – proxyphylline, xanthinol, and diprophylline employing various recombinant short-chain ADHs with (R)- or (S)-selectivity as key biocatalysts. By choosing the appropriate ADH, the (R)- as well as the (S)-enantiomer of proxyphylline was prepared in excellent enantiomeric excess (99–99.9% ee), >99% conversion, and the isolated yield ranging from 65% to 74%, depending on the used biocatalyst (ADH-A from Rhodococcus ruber or a variant derived from Lactobacillus kefir, Lk-ADH-Lica). In turn, E. coli/ADH-catalyzed bioreduction of the carbonylic precursor of xanthinol and diprophylline furnished the corresponding (S)-chlorohydrin in >99% ee, >99% conversion, and 80% yield (in the case of Lk-ADH-Lica); while the (R)-counterpart was afforded in 94% ee, 64% conversion, and 41% yield (in the case of SyADH from Sphingobium yanoikuyae). After further chemical functionalization of the key (S)-chlorohydrin intermediate, the desired homochiral (R)-xanthinol (>99% ee) was obtained in 97% yield and (S)-diprophylline (>99% ee) in 90% yield. The devised biocatalytic method is straightforward and thus might be considered practical in the manufacturing of title pharmaceuticals.
31. Chemoenzymatic deracemization of lisofylline catalyzed by a (laccase/TEMPO)-alcohol dehydrogenase system
P. Borowiecki,* A. Rudzka, T. Reiter, W. Kroutil
Catal. Sci. Technol. 2022, 12, 4312–4324.
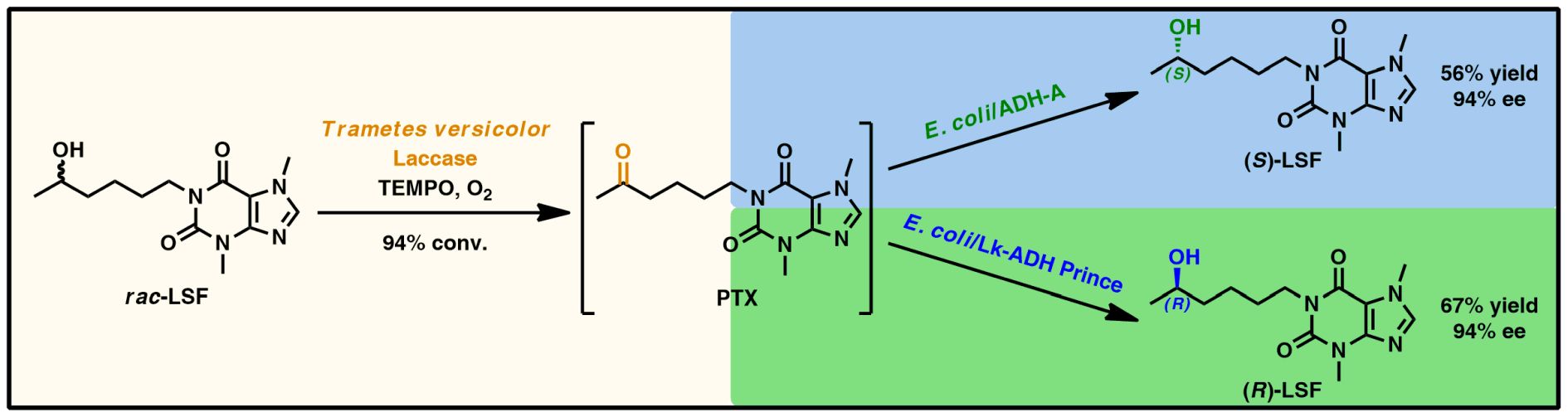
Lisofylline (LSF) is a synthetic methylxanthine active agent exhibiting potent anti-inflammatory and immunomodulatory properties; therefore, it has been widely investigated as a promising drug candidate for treating various autoimmune disorders, including type 1 diabetes. In this study, we report on developing a sequential chemoenzymatic one-pot two-step deracemization protocol for racemic LSF. This task was accomplished in a stereo-complementary manner via a tandem bi-enzymatic oxidation–reduction reaction sequence composed of (i) non-selective chemoenzymatic aerobic oxidation of LSF to pentoxifylline (PTX) catalyzed by commercially available laccase from Trametes versicolor (LTv) and 2,2,6,6-tetramethylpiperidinyloxy radical (TEMPO) as a redox mediator, and (ii) stereoselective bioreduction of in situ formed PTX to give enantiomeric LSF, which was catalyzed by home-made lyophilized E. coli cells harboring overexpressed alcohol dehydrogenases (ADHs) with complementary stereospecificity. Firstly, a multi-step optimization procedure of LTv/TEMPO-catalyzed oxidation of LSF allowed achieving dramatic improvement of the conversion rates from an initial 16% up to 95%, demonstrating the high synthetic potency of this method compared to traditional chemical reactions requiring toxic oxidants used in stoichiometric amounts. In turn, separate stereoselective bioreductions of PTX using recombinant ADHs from Rhodococcus ruber (E. coli/ADH-A) and Lactobacillus kefiri (E. coli/LK-ADH Prince) furnished both LSF enantiomers (>99% ee) with high 93–94% conversion and in 65–67% yield range, respectively. The coupling of the above-mentioned chemoenzymatic steps afforded both antipodes of LSF on a preparative scale (0.16 mmol of racemic LSF) in the range of 56–67% yield and 94% ee depending on the employed ADHs.
doi: 10.1039/d2cy00145d
30. Expanding Access to Optically Active Non-Steroidal Anti-Inflammatory Drugs via Lipase-Catalyzed KR of Racemic Acids Using Trialkyl Orthoesters as Irreversible Alkoxy Group Donors
B. Zdun, P. Cieśla, J. Kutner, P. Borowiecki*
Catalysts 2022, 12, 546–566.
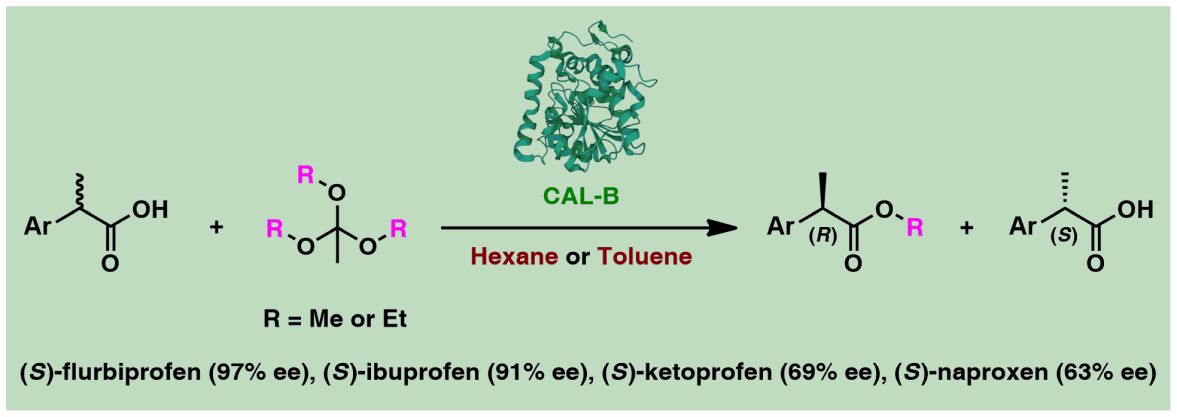
Studies into the enzymatic kinetic resolution (EKR) of 2-arylpropanoic acids (‘profens’), as the active pharmaceutical ingredients (APIs) of blockbuster non-steroidal anti-inflammatory drugs (NSAIDs), by using various trialkyl orthoesters as irreversible alkoxy group donors in organic media, were performed. The enzymatic reactions of target substrates were optimized using several different immobilized preparations of lipase type B from the yeast Candida antarctica (CAL-B). The influence of crucial parameters, including the type of enzyme and alkoxy agent, as well as the nature of the organic co-solvent and time of the process on the conversion and enantioselectivity of the enzymatic kinetic resolution, is described. The optimal EKR procedure for the racemic profens consisted of a Novozym 435-STREM lipase preparation suspended in a mixture of 3 equiv of trimethyl or triethyl orthoacetate as alkoxy donor and toluene or n-hexane as co-solvent, depending on the employed racemic NSAIDs. The reported biocatalytic system provided optically active products with moderate-to-good enantioselectivity upon esterification lasting for 7–48 h, with most promising results in terms of enantiomeric purity of the pharmacologically active enantiomers of title APIs obtained on the analytical scale for: (S)-flurbiprofen (97% ee), (S)-ibuprofen (91% ee), (S)-ketoprofen (69% ee), and (S)-naproxen (63% ee), respectively. In turn, the employment of optimal conditions on a preparativescale enabled us to obtain the (S)-enantiomers of: flurbiprofen in 28% yield and 97% ee, ibuprofen in 45% yield and 56% ee, (S)-ketoprofen in 23% yield and 69% ee, and naproxen in 42% yield and 57% ee, respectively. The devised method turned out to be inefficient toward racemic etodolac regardless of the lipase and alkoxy group donor used, proving that it is unsuitable for carboxylic acids possessing tertiary chiral centers.
2021
29. From Waste to Value—Direct Utilization of α-Angelica Lactone as a Nonconventional Irreversible Acylating Agent in a Chromatography-Free Lipase-Catalyzed KR Approach toward sec-Alcohols
M. Poterała, P. Borowiecki*
ACS Sustain. Chem. Eng. 2021, 9, 10276-10290.
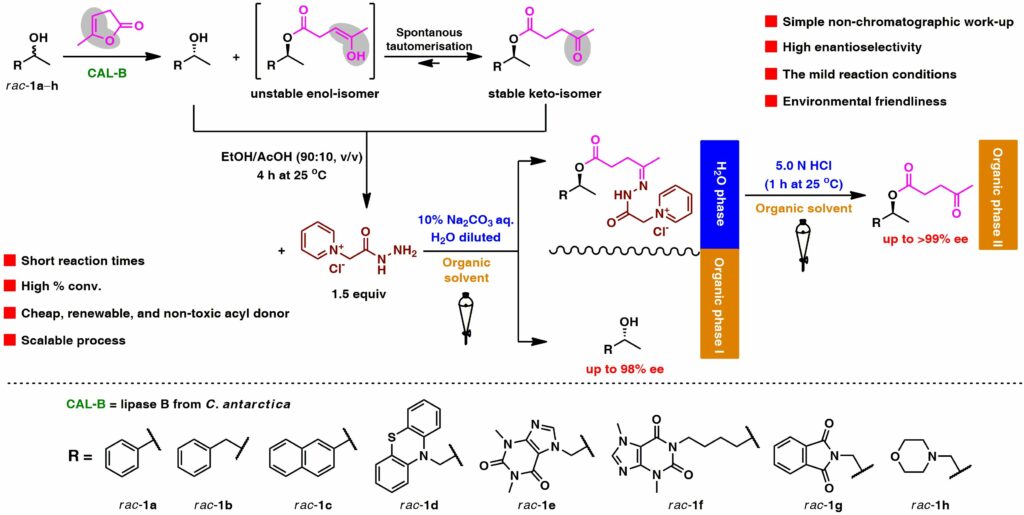
Although the classical enzymatic kinetic resolutions (EKRs) are among the most important reactions in biocatalysis, the requirement of application of chromatographic purification technique, which is responsible for the generation of large amounts of waste organic solvents, is its major drawback. To minimize the environmental impact of such attempts and to address the current sustainability challenges, we decided to develop a novel EKR methodology, which relies on the usage of cheap, fully renewable, nontoxic, and irreversible acyl group donor reagent, namely, α-angelica lactone. The employed activated enol lactone-based acyl donor proved to be a versatile reagent enabling efficient and highly enantioselective (up to E ≫ 500) lipase-catalyzed resolution of a set of racemic sec-alcohols with up to >99% ee and near to quantitative isolation yields according to conversions achieved during the respective EKR procedure. Moreover, α-angelica lactone provided, in this case, an ability of fast and straightforward separation of the enzymatic reactions’ products via chromatography-free reactive liquid−liquid extraction (LLE) workup using either saturated aqueous sodium bisulfate in dimethylformamide (DMF) or Girard’s P reagent in a mixture of EtOH/AcOH (90:10, v/v), respectively. The NaHSO3/DMF LLE workup and the subsequent reisolation of the respective levulinate from the aqueous layer turned out to be less efficient than a separation with Girard’s P reagent. The selective LLE of optically active levulinate-Girard P-hydrazones from the respective alcohols and further hydrolysis of the respective hydrazides with diluted HClaq. could be performed with high levels of recovery of both EKR products isolated usually without loss of enantiomeric purity.
28. Synthesis of Novel Acyl Derivatives of 3‐(4,5,6,7‐Tetrabromo‐1H‐benzimidazol‐1‐yl)propan‐1‐ols — Intracellular TBBi‐Based CK2 Inhibitors with Proapoptotic Properties
K. Chojnacki, P. Wińska,* O. Karatsai, M. Koronkiewicz, M. Milner‐Krawczyk, M. Wielechowska, M. J. Rędowicz, M. Bretner, P. Borowiecki
Int. J. Mol. Sci. 2021, 22, 6261–6283.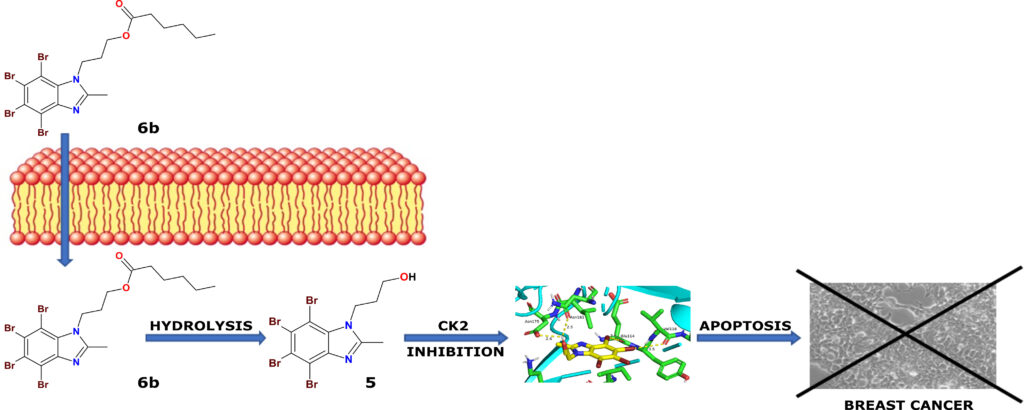
Protein kinase CK2 has been considered an attractive drug target for anti‐cancer therapy. The synthesis of N‐hydroxypropyl TBBi and 2MeTBBi derivatives as well as their respective esters was carried out by using chemoenzymatic methods. Concomitantly with kinetic studies toward recombinant CK2, the influence of the obtained compounds on the viability of two human breast carcinoma cell lines (MCF‐7 and MDA‐MB‐231) was evaluated using MTT assay. Additionally, an intracellular inhibition of CK2, as well as an induction of apoptosis in the examined cells after the treatment with the most active compounds, were studied by Western blot analysis, phase-contrast microscopy, and flow cytometry method. The results of the MTT test revealed potent cytotoxic activities for most of the newly synthesized compounds (EC50 4.90 to 32.77 μM), corresponding to their solubility in biological media. We concluded that derivatives with the methyl group decrease the viability of both cell lines more efficiently than their non‐methylated analogs. Furthermore, inhibition of CK2 in breast cancer cells treated with the tested compounds at concentrations equal to their EC50 values correlates well with their lipophilicity since derivatives with higher values of logP are more potent intracellular inhibitors of CK2 with better proapoptotic properties than their parental hydroxyl compounds.
doi: 10.3390/ijms22126261
27. The First Insight Into the Supramolecular System of D,L-α-Difluoromethylornithine: A New Antiviral Perspective
J. Bojarska*, R. New, P. Borowiecki, M. Remko, M. Breza, I. D. Madura, A. Fruziński, A. Pietrzak, W. M. Wolf
Front. Chem. 2021, 9, 679776–679799.
Targeting the polyamine biosynthetic pathway by inhibiting ornithine decarboxylase (ODC) is a powerful approach in the fight against diverse viruses, including SARS-CoV-2. Difluoromethylornithine (DFMO, eflornithine) is the best-known inhibitor of ODC and a broad spectrum, unique therapeutical agent. Nevertheless, its pharmacokinetic profile is not perfect, especially when large doses are required in antiviral treatment. This article presents a holistic study focusing on the molecular and supramolecular structure of DFMO and the design of its analogues toward the development of safer and more effective formulations. In this context, we provide the first deep insight into the supramolecular system of DFMO supplemented by a comprehensive, qualitative and quantitative survey of non-covalent interactions via Hirshfeld surface, molecular electrostatic potential, enrichment ratio and energy frameworks analysis visualizing 3-D topology of interactions in order to understand the differences in the cooperativity of interactions involved in the formation of either basic or large synthons (Long-range Synthon Aufbau Modules, LSAM) at the subsequent levels of well-organized supramolecular self-assembly, in comparison with the ornithine structure. In the light of the drug discovery, supramolecular studies of amino acids, essential constituents of proteins, are of prime importance. In brief, the same amino-carboxy synthons are observed in the bio-system containing DFMO. DFT calculations revealed that the biological environment changes the molecular structure of DFMO only slightly. The ADMET profile of structural modifications of DFMO and optimization of its analogue as a new promising drug via molecular docking are discussed in detail.
26. Chemoenzymatic Enantioselective and Stereo-Convergent Syntheses of Lisofylline Enantiomers via Lipase-catalyzed Kinetic Resolution and Optical Inversion Approach
P. Borowiecki,* B. Zdun, M. Dranka
Mol. Catal. 2021, 504, 111451–111470.
Highly enantioselective enzymatic kinetic resolution (EKR) of racemic lisofylline is presented for the first time. A comprehensive optimization of the key parameters of lipase-catalyzed transesterification of racemic lisofylline revealed that optimal biocatalytic system consisted of immobilized lipase type B from Candida antarctica (Chirazyme L-2, C-3) suspended in a mixture of 3 equiv of vinyl acetate as an acetyl donor and ethyl acetate as a solvent. Under optimal reaction conditions, the 1 gram-scale (Chirazyme L-2, C-3)-catalyzed kinetic resolution of racemic lisofylline furnished both the EKR products in a homochiral form (>99% ee) with the 50% conv., and the highest possible enantioselectivity.
The best results in terms of the reaction yields (47–50%) and enantiomeric purity of the kinetically-resolved optically active products were achieved when the preparative-scale EKR was carried out for 2 h at 60 ºC. In addition, stereoinversion of the less biologically-relevant (S)-lisofylline into its (R)-enantiomer was successfully achieved via acetolysis of the respective optically pure (S)-mesylate by using 2 equiv of ceasium acetate and catalytic amount of 18-Crown-6 in dry toluene, followed by K2CO3-mediated methanolysis of (R)-acetate. The elaborated EKR methodology together with enantioconvergent strategy provided a useful chemoenzymatic protocol for the synthesis of complementary enantiomers of titled anti-inflammatory API.
Moreover, we report on the first single-crystal X-ray diffraction (XRD) analyses performed for the synthesized lisofylline enantiomers. Insight into the source of CAL-B stereoselectivity toward racemic lisofylline was gained by molecular docking experiments. In silico theoretical predictions matched very well with experimental results.
25. Chemoenzymatic Synthesis of Enantiomerically Enriched Diprophylline and Xanthinol Nicotinate
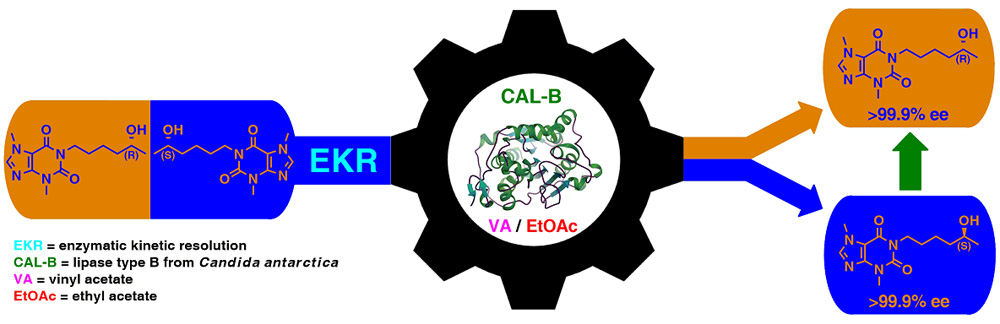
P. Borowiecki,* M. Młynek, M. Dranka
Bioorg. Chem. 2021, 106, 104448–104464.
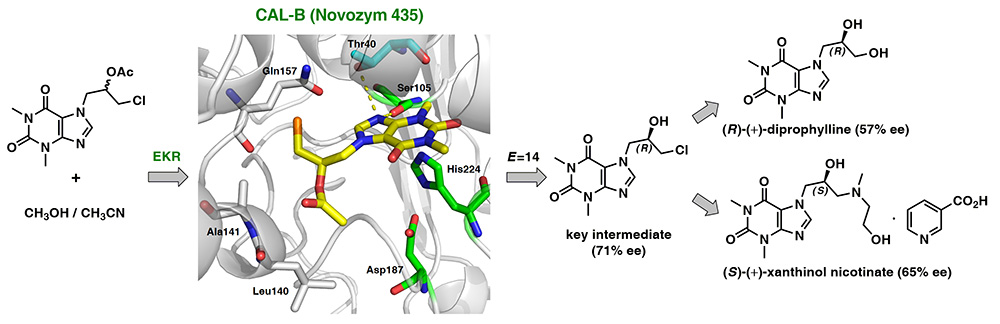
A concise chemoenzymatic route toward enantiomerically enriched active pharmaceutical ingredients (API) – diprophylline and xanthinol nicotinate – is reported for the first time. The decisive step is an enantioselective lipase-mediated methanolysis of racemic chlorohydrin-synthon acetate, namely 1-chloro-3-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)propan-2-yl acetate, performed under kinetically-controlled conditions on a preparative 500 mg-scale. The best results in terms of reaction enantioselectivity (E=14) were obtained for the enantiomers resolution performed with lipase type B from Candida antarctica immobilized on acrylic resin (CAL-B, Novozym 435) suspended in homo-phasic acetonitrile-methanol mixture.
The elaborated biocatalytic system furnished the key chlorohydrin intermediate (in 71% ee and 38% yield), which was then smoothly converted into enantioenriched active agents: (R)-(–)-diprophylline (57% ee) and (S)-(+)-xanthinol nicotinate (65% ee). To support the assignment of absolute configurations of EKR-products as well as to confirm the stereochemical outcome of the remaining reaction steps, docking studies toward the prediction of enantiomers binding selectivity in CAL-B active site as well as the respective chemical correlations with enantiomerically enriched analytical standards obtained from commercially available (R)-(–)-epichlorohydrin, were applied. In addition, single-crystal X-ray diffraction (XRD) analyses were performed for the synthesized optically active APIs furnishing by this manner a first crystal structures of nicotinic acid salt of xanthinol.
2020
24. Antifungal Polybrominated Proxyphylline Derivative Induces Candida albicans Calcineurin Stress Response in Galleria mellonella
M. Gizińska, A. Staniszewska, M. Kazek, M. Koronkiewicz, Ł. Kuryk, M. Milner-Krawczyk, J. Baran, P. Borowiecki,* M. Staniszewska*
Bioorg. Med. Chem. Lett. 2020, 30, 127545–127550.
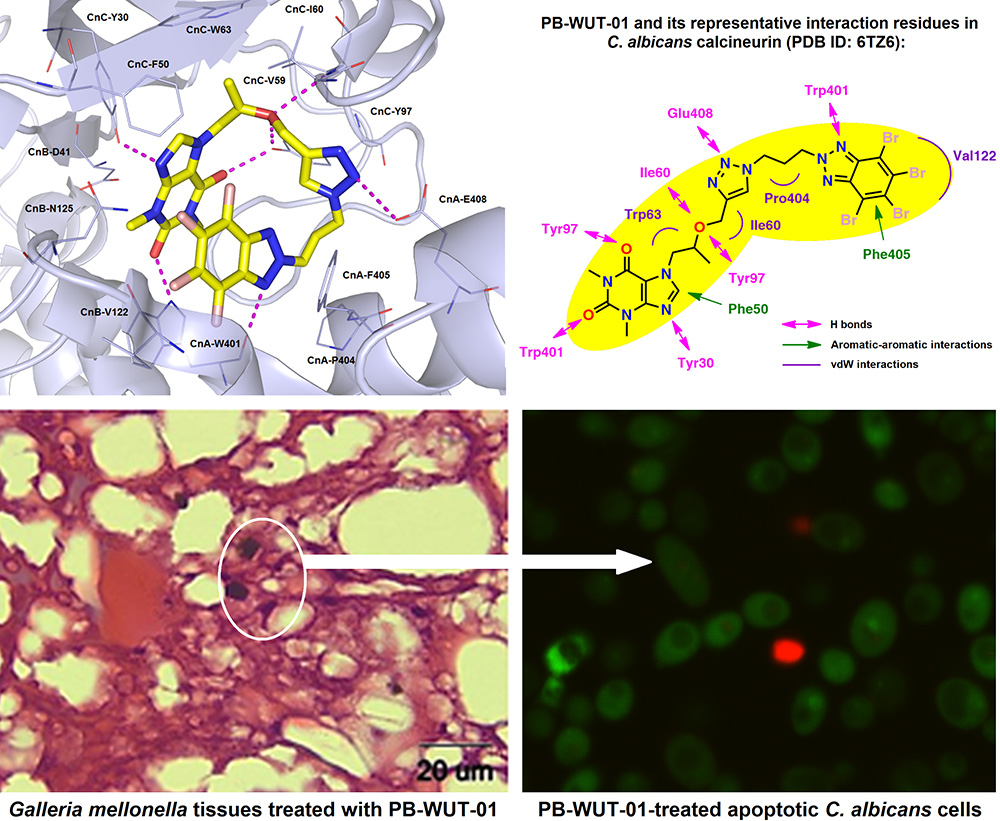
Candida albicans CNB1 plays a role in the response in vitro and in vivo to stress generated by PB-WUT-01, namely 1,3-dimethyl-7-(2-((1-(3-(perbromo-2H-benzo[d][1,2,3]triazol-2-yl)propyl)-1H-1,2,3-triazol-4-yl)methoxy) propyl)-1H-purine-2,6(3H,7H)-dione. The antifungal mechanism involved the calcineurin pathway-regulated genes SAP9-10. Galleria mellonella treated with PB-WUT-01 (at 0.64 μg/mg) showed limited candidiasis and remained within the highest survival rates. The molecular mode of action of PB-WUT-01 was rationalized by in silico docking studies toward both human and C. albicans calcineurin A (CNA) and calcineurin B (CNB) complexes, respectively. PB-WUT-01 acting as a calcineurin inhibitor in the C. albicans cells enhances the cells’ susceptibility. Therefore, it could be a suitable alternative treatment in patients with candidiasis.
23. Biocatalytic Asymmetric Reduction of γ-Keto Esters to Access Optically Active γ-Aryl-γ-butyrolactones
P. Borowiecki,* N. Telatycka, M. Tataruch, A. Żądło-Dobrowolska, T. Reiter, K. Schühle, J. Heider, M. Szaleniec, W. Kroutil
Adv. Synth. Catal. 2020, 362, 2012–2029.
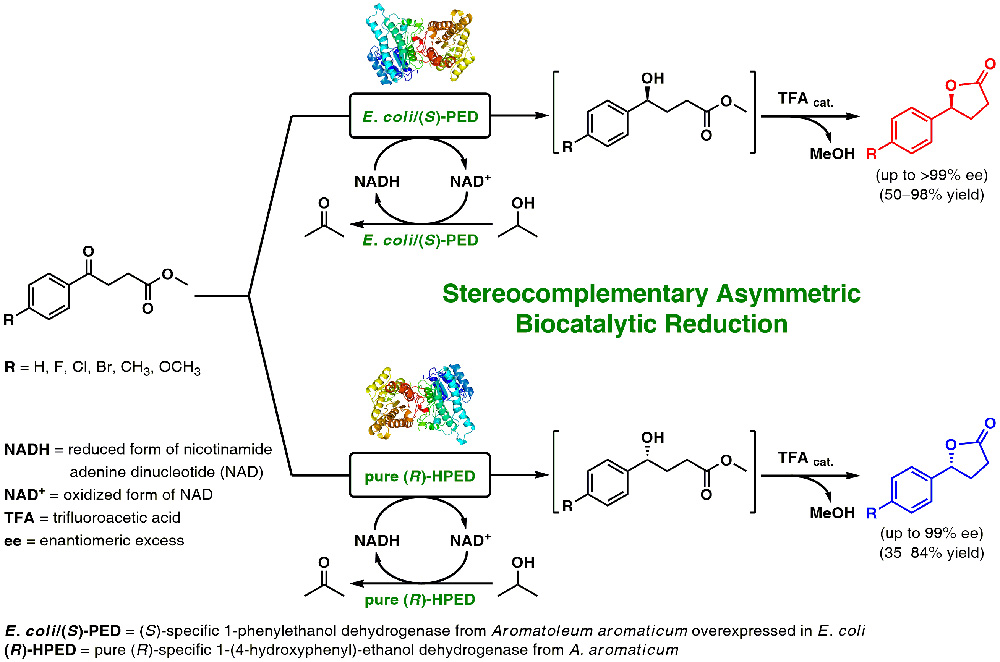
An efficient stereoselective syntheses of a series of functionalized optically active γ-aryl-γ-butyrolactones is achieved by enzymatic asymmetric reduction of the corresponding sterically demanding γ-keto esters employing wild-type and recombinant alcohol dehydrogenases. The best stereoselectivities for the reduction via hydrogen transfer was obtained with two short chain dehydrogenases (SDRs) of complementary stereospecificity from Aromatoleum aromaticum, namely the Prelog-specific NADH-dependent (S)-1-phenylethanol dehydrogenase [(S)-PED] and the anti-Prelog-specific (R)-1-(4-hydroxyphenyl)-ethanol dehydrogenase [(R)-HPED], respectively. Biotransformations catalyzed by both enzymes, followed by TFA-catalyzed cyclization of the resulting γ-hydroxy esters, furnished the respective (S)- and (R)-configured products with exquisite optical purity (up to >99% ee).
The synthetic value was demonstrated on preparative scale for the asymmetric bioreduction of the model compound, methyl 4-oxo-4-phenylbutanoate, affording optically pure (S)-γ-phenyl-γ-butyrolactone (>99% ee) in 67–74% isolated yield at 89–95% conversion depending on the applied scale.
2019
22. Highly Efficient, Solvent-free Esterification of Testosterone Promoted by a Recyclable Polymer-supported Tosylic Acid Catalyst Under Microwave Irradiation
P. Borowiecki,* M. Kraszewski
ARKIVOC 2019, (vi), 288–305.
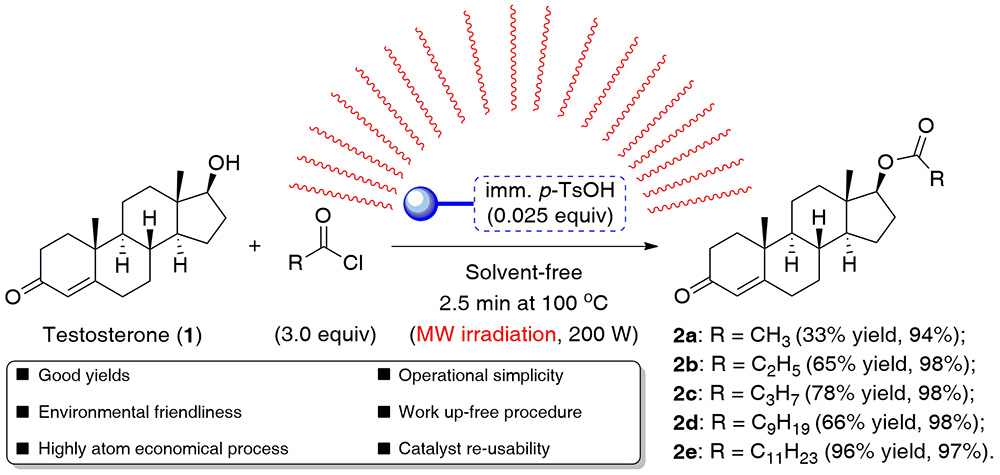 Although the classical acylation of testosterone clearly benefits from a broad substrate scope and available catalysts, the requirement of hazardous reagents and the high waste production are its drawbacks. To optimize the process efficiency as well as minimize the environmental impact, we decided to develop a novel method of testosterone esters synthesis, which relies on the usage of recyclable heterogeneous polymer-supported tosylic acid catalyst and microwave-assistance effect in a non-solvent system. Under the established MW-conditions, the acceleration of the process rate was so efficient that the reaction completed within 2.5 min, thus affording the desired esters in the 33–96% yield range without using a work-up procedure.
Although the classical acylation of testosterone clearly benefits from a broad substrate scope and available catalysts, the requirement of hazardous reagents and the high waste production are its drawbacks. To optimize the process efficiency as well as minimize the environmental impact, we decided to develop a novel method of testosterone esters synthesis, which relies on the usage of recyclable heterogeneous polymer-supported tosylic acid catalyst and microwave-assistance effect in a non-solvent system. Under the established MW-conditions, the acceleration of the process rate was so efficient that the reaction completed within 2.5 min, thus affording the desired esters in the 33–96% yield range without using a work-up procedure.
Furthermore, the elaborated catalytic system could be recycled for at least 2 runs not only without a loss of the products yield, but unexpectedly with significant improvement of the reaction efficiency, which may indicate that the reduction of the catalyst loading is possible. We believe that this finding constitutes a very good starting-point for further optimization of the studied process.
21. A facile Lipase-catalyzed KR Approach Toward Enantiomerically Enriched Homopropargyl Alcohols
P. Borowiecki,* M. Dranka
Bioorganic Chemistry 2019, 93, 102754–102765.
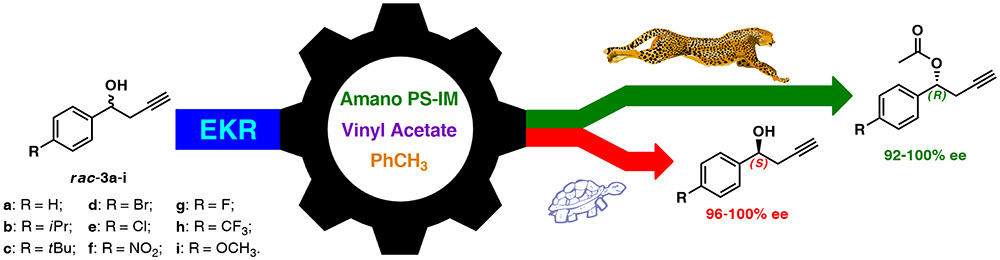
Compounds possessing propargylic (prop-2-ynylic) system are very important building blocks for organic chemistry. Among them, preparation of enantiomeric homopropargyl alcohols (but-3-yn-1-ols) constitutes a key challenge for asymmetric synthesis and thus drawn tremendous attention from the synthetic community in the last few decades. In this work, the catalytic performance of a set of commercial lipases has been investigated for enantioselective transesterification of 1-phenylhomopropargylic alcohols under kinetically-controlled conditions. Lipase from Burckholderia cepacia (BCL) immobilized either on ceramic (Amano PS-C II) or diatomaceous earth (Amano PS-IM) turned out to be the most active and enantioselective enzyme preparations (E≫500) furnishing both resolution products of the racemic 1-phenylbut-3-yn-1-ol in highly enantiomerically enriched form (up >99% ee).
Variable reaction parameters, such as the acyl-group donor reagent as well as solvent, were additionally screened to establish their impact on the stereochemical outcome. For optimal biocatalytic systems established with model substrate, the enzymatic transformations were extended toward preparative scale KR of 8 other differently para-phenyl-substituted homopropargylic sec-alcohols, which resulted in the synthesis of (S)-alcohols (96–100% ee) and the respective (R)-acetates (92–100% ee) in 19–44% yield, accordingly. Additionally, the crystal structure of (1R)-1-(4-nitrophenyl)but-3-yn-1-yl acetate has been evaluated for the first time and helped to assess stereopreference of the studied BCL.
20. New Applications of Biotechnology in the Field of Pharmaceutical Syntheses
G. Grynkiewicz,* P. Borowiecki
Przemysł Chemiczny 2019, 98/3, 434–441.

There is a significant increase in application of biotransformation as a crucial step in API syntheses, in which chirality center of the proper configuration is installed, in recent decades. Initial applications of hydrolytic enzymes including mostly lipases, proteases and acylases for separation of racemic pharmaceutical key-intermediates and APIs, were more recently augmented by biocatalysts capable of stereoselective hydrogenation of prochiral ketones or olefins, greatly expanding the field of biocatalytic pharmaceutical syntheses. Examples of new processes for APIs featuring α-, β-, and γ-amino acid structures (L-DOPA; sitagliptin; pregabalin) are presented to illustrate the point. Remarkable progress in directed evolution of enzymatic catalysts (honored by Nobel prize in chemistry for 2018) designed for specific needs of industrial processes is likely to influence seriously strategies of API development and manufacturing.
2018
19. Synthesis of Novel Proxyphylline Derivatives with dual Anti-Candida albicans and Anticancer activity
P. Borowiecki,* P. Wińska, M. Bretner, M. Gizińska, M. Koronkiewicz, M. Staniszewska*
European Journal of Medicinal Chemistry 2018, 150, 307–333.
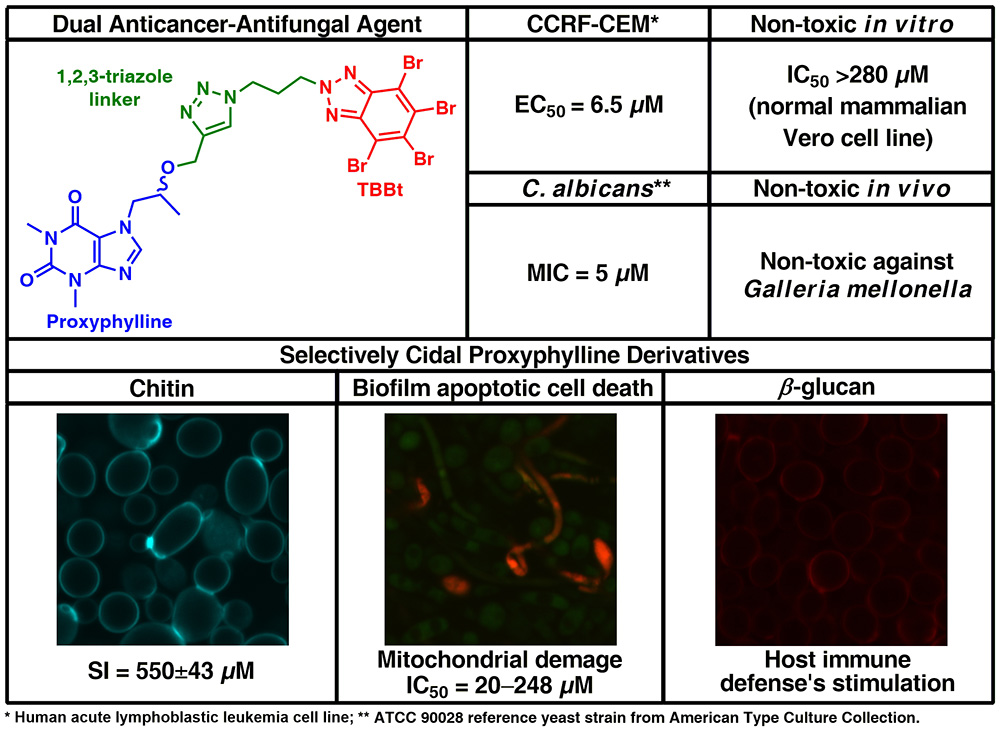
Three out of 16 newly synthesized 1,3-dimethylxanthine derivatives (proxyphylline analogues) exhibited consistencies between antifungal and anticancer properties. Proxyphylline possessing 1-(10H-phenothiazin-10-yl)propan-2-yl (6) and polybrominated benzimidazole (41) or benzotriazole moiety (42) remained selectively cidal against Candida albicans (lg R≥3 at conc. of 31, 36 and 20 μM, respectively) however not against normal mammalian Vero cell line in vitro (IC50≥280 μM) and Galleria mellonella in vivo. These compounds also displayed moderate antineoplastic activity against human breast adenocarcinoma (MCF-7) cell line (EC50=80 μM) and high against peripheral blood T lymphoblast (CCRF-CEM) (EC50=6.3–6.5 μM). In addition, 6 and 42 exerted: (1) dual activity against fungal adhesion and damage mature biofilm; (2) necrosis of planktonic cells due to loss of membrane function and of structural integrity; (3) biochemical (inhibition of sessile cell respiration) and morphological changes in cell wall polysaccharide contents. Therefore, leading proxyphylline derivatives can be employed to prevent cancer-associated biofilm Candida infections.
2017
18. Lipase-catalyzed Kinetic Resolution Approach Toward Enantiomerically Enriched 1-(β-Hydroxypropyl)indoles
P. Borowiecki,* I. Justyniak, Z. Ochal
Tetrahedron Asymm. 2017, 28, 1717–1732.
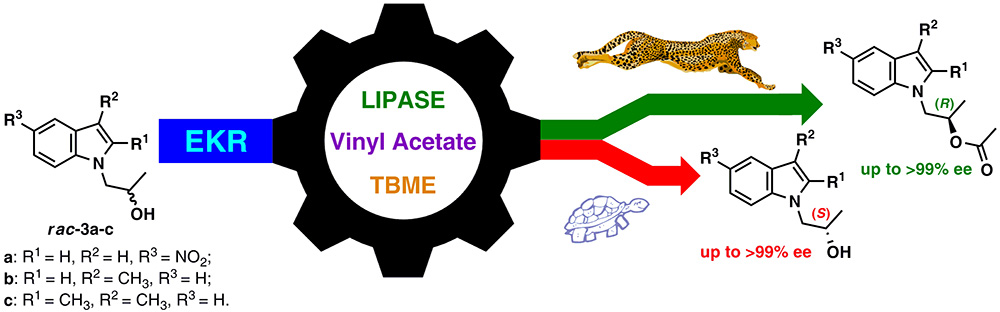
In a route towards enantiomerically enriched 1-(β-hydroxypropyl)indoles, which are potentially useful building blocks for high value-added chemicals synthesis, a kinetic resolution approach by means of lipase-catalyzed enantioselective acylation as well as hydrolysis/methanolysis has been elaborated for the first time. The enzymatic resolution of chiral N-substituted indole-based sec-alcohols was successfully accomplished, yielding both enantiomeric forms of the employed derivatives with up to >99% optical purity via an enantioselective transesterification under mild reaction conditions.
The most selective resolutions were obtained using fungal (CAL-B and TLL) and bacterial (PFL and BCL) lipases and vinyl acetate as the acyl group donor. The synthetic protocol described herein is very simple, user-friendly and efficient, thus paving the way for future access toward more complex compounds of this type. Absolute configuration of novel enantiomeric derivatives, and thus stereochemistry of the described enzymatic reactions was confirmed by application of CDA-based NMR methodology and single-crystal X-ray diffraction analysis.
17. Lipase‐Catalyzed Kinetic Resolution of N‐Substituted 1‐(β‐Hydroxypropyl)indoles by Enantioselective Acetylation
P. Borowiecki,* M. Dranka; Z. Ochal
European Journal of Organic Chemistry 2017, 2017, 5378–5390.
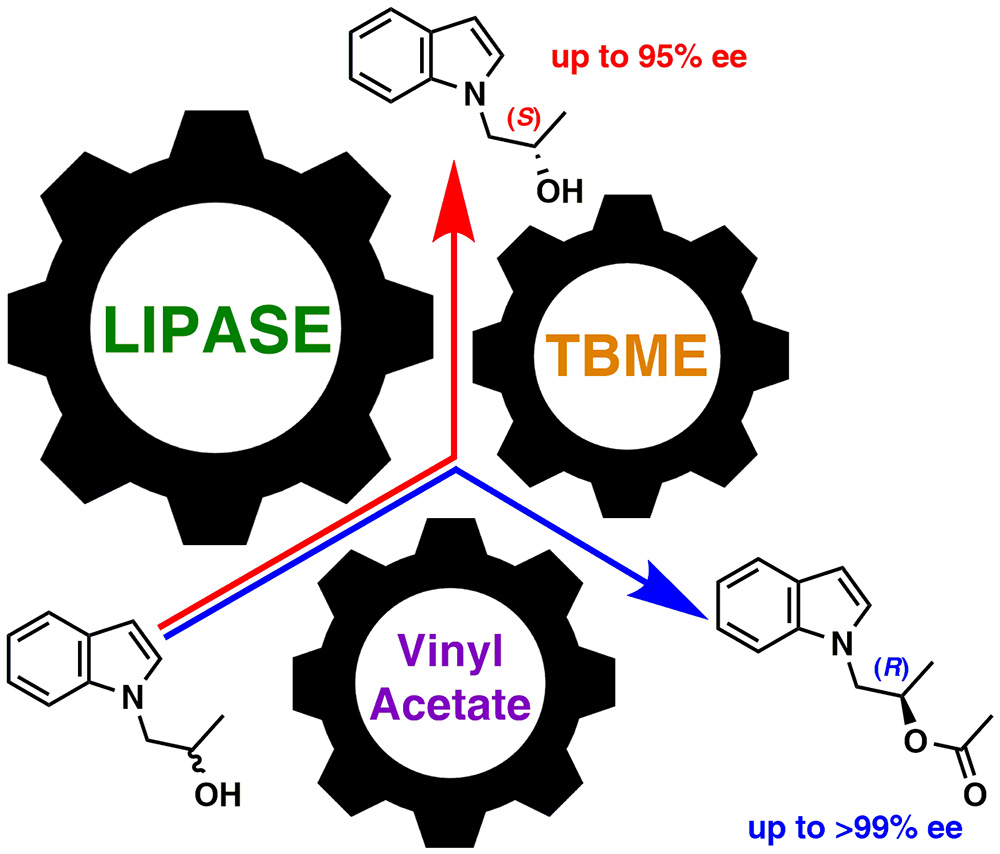
A novel, concise and efficient route towards both antipodes of N-(β-hydroxypropyl)-indoles is described. The key-step is an enantioselective lipase-mediated acetylation of racemic indolic alcohols [1-(1H-indol-1-yl)propan-2-ol, 1-(2-methyl-1H-indol-1-yl)propan-2-ol and 1-(5-bromo-1H-indol-1-yl)propan-2-ol] with enol esters (vinyl acetate and isopropenyl acetate) as an acyl group donor performed under kinetically–controlled conditions. A substantial influence of enzyme type, acyl group donor, and organic solvent on conversion rates and enantioselectivity of the enzymatic kinetic resolutions (EKRs) of respective racemates were examined in detail.
The best results in terms of reaction enantioselectivity were obtained for the resolutions performed with native lipase from Pseudomonas fluorescens (PFL, Amano AK) in diisopropyl ether (DIPE), or tert-butyl methyl ether (TBME) solution. To support the assignment of the absolute configurations of EKR-products chemical correlations with enantiomerically enriched analytical standards and single-crystal X-ray diffraction analysis were applied.
16. Chemoenzymatic Preparation of Enantiomerically Enriched (R)-(–)-Mandelic Acid Derivatives: Application in the Synthesis of the Active Agent Pemoline
M. Poterała,* M. Dranka, P. Borowiecki*
European Journal of Organic Chemistry 2017, 16, 2290–2304.
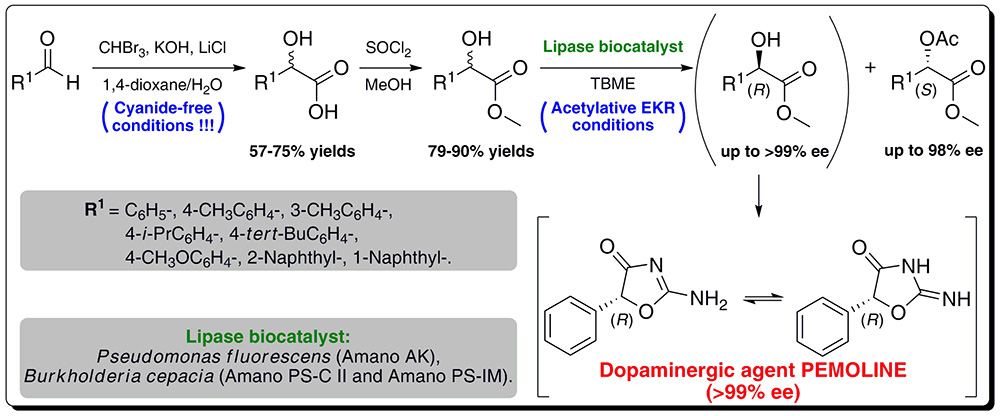
The enantioselective resolutions of a several racemic derivatives of mandelic acid methyl ester catalyzed by lipases from Pseudomonas fluorescens (Amano AK) or Burkholderia cepacia (Amano PS-C II and Amano PS-IM) are demonstrated. A gram-scale lipase-mediated kinetic resolution approach have been developed, allowing facile synthesis of the corresponding (R)-(–)-methyl mandelates with excellent enantiomeric excesses (up to >99% ee) and the reactions enantioselectivity (E-value up to >200).
The dopaminergic agent pemoline – used in the treatment of attention-deficit hyperactivity disorder (ADHD) and narcolepsy – was synthesized with 98% ee in a straightforward route by condensing the prepared methyl (R)-(–)-mandelate with guanidine hydrochloride under basic conditions. The desired (R)-(+)-pemoline in an optically pure form (>99% ee) was obtained after two recrystallizations from ethanol. However, it was confirmed by chiral HPLC that optically active pemoline undergoes racemization in methanol solution.
2016
15. Chemoenzymatic Synthesis of Proxyphylline Enantiomers
P. Borowiecki,* D. Paprocki, A. Dudzik, J. Plenkiewicz
The Journal of Organic Chemistry 2016, 81, 380–395.
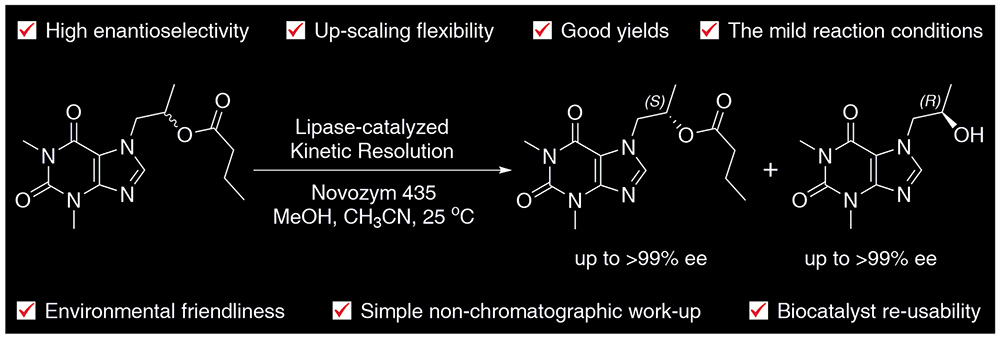
A novel synthetic route for preparation of proxyphylline enantiomers using a kinetic resolution (KR) procedure as the key step is presented. The reactions were catalyzed by immobilized Candida antarctica lipase B in acetonitrile. Three types of reactions were examined: (i) enantioselective transesterification of racemic proxyphylline with vinyl acetate as well as (ii) hydrolysis and (iii) methanolysis of its esters. The influence of reaction conditions on the substrate conversion and enantiomeric purity of the products were investigated. Studies on analytical scale reactions revealed that the titled API enantiomers could be successfully obtained with excellent enantiomeric excess (up to >99% ee).
The process was easily conducted on a 5 g scale at 100 g/L. In a preparative-scale reaction, unreacted (S)-(+)-butanoate (97% ee) and (R)-(–)-alcohol (96% ee) were obtained after 2 days in yields of 45% and 46%, respectively. When the reaction time was extended to 6 days (S)-(+)-butanoate was isolated in >99% ee and acceptable high enantioselectivity (E=90). Importantly, the KR’s products could be conveniently isolated by exploiting different solubility of the ester/alcohol in acetonitrile at room temperature. In addition, a chiral preference of the CAL-B active site for the R-enantiomer was rationalized by in silico docking studies.
2015
14. Asymmetric Reduction of Ketones and β-Keto Esters by (S)-1-Phenylethanol Dehydrogenase from Denitrifying Bacterium Aromatoleum aromaticum
A.. Dudzik, W. Snoch, P. Borowiecki, J. Opalinska-Piskorz, M. Witko, J. Heider, M. Szaleniec*
Applied Microbiology and Biotechnology 2015, 99, 5055–5069.
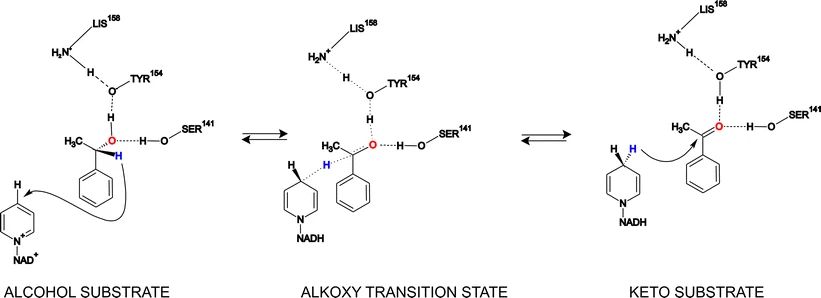
Enzyme-catalyzed enantioselective reductions of ketones and keto esters have become popular for the production of homochiral building blocks which are valuable synthons for the preparation of biologically active compounds at industrial scale. Among many kinds of biocatalysts, dehydrogenases/reductases from various microorganisms have been used to prepare optically pure enantiomers from carbonyl compounds.
(S)-1-Phenylethanol dehydrogenase (PEDH) was found in the denitrifying bacterium Aromatoleum aromaticum (strain EbN1) and belongs to the short-chain dehydrogenase/reductase family. It catalyzes the stereospecific oxidation of (S)-1-phenylethanol to acetophenone during anaerobic ethylbenzene mineralization, but also the reverse reaction, i.e., NADH-dependent enantioselective reduction of acetophenone to (S)-1-phenylethanol. In this work, we present the application of PEDH for asymmetric reduction of 42 prochiral ketones and 11 β-keto esters to enantiopure secondary alcohols.
The high enantioselectivity of the reaction is explained by docking experiments and analysis of the interaction and binding energies of the theoretical enzyme-substrate complexes leading to the respective (S)- or (R)-alcohols. The conversions were carried out in a batch reactor using Escherichia coli cells with heterologously produced PEDH as whole-cell catalysts and isopropanol as reaction solvent and co-substrate for NADH recovery. Ketones were converted to the respective secondary alcohols with excellent enantiomeric excesses and high productivities.
Moreover, the progress of product formation was studied for nine para-substituted acetophenone derivatives and described by neural network models, which allow to predict reactor behavior and provides insight on enzyme reactivity. Finally, equilibrium constants for conversion of these substrates were derived from the progress curves of the reactions. The obtained values matched very well with theoretical predictions.
13. Enantiodifferentiation of Promethazine using (S)-(–)-BINOL as the NMR Chiral Solvating Agent – Determination of the Enantiomeric Purity and Performance comparison with Traditional Chiral HPLC
P. Borowiecki*
Tetrahedron: Asymmetry 2015, 26, 16–23.
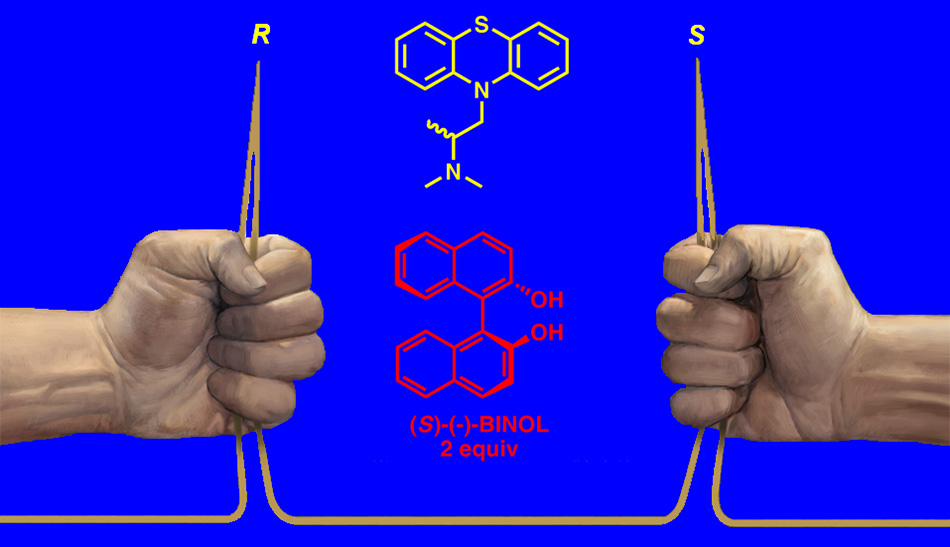
Enantiomeric excess of optically active promethazine was determined for the first time by proton nuclear magnetic resonance (1H NMR) spectroscopic enantiodifferentiation method by means of (S)-(–)-1,1′-Bi(2-naphthol) [(S)-(–)-BINOL] as chiral solvating agent (CSA), and additionally ascertained by HPLC measurements on Chiralcel® OJ chiral column. The obtained results for enantiopurity of standard samples of promethazine were almost fully consistent with those established by chiral HPLC (quantitative to approximately 1% minor enantiomer). Developed method is simple, rapid, inexpensive, repeatable, sensitive, and very selective toward promethazine enantiomers, and may serve for its routine quantitative analysis.
12. Industrial Applications of Lipases in the Synthesis of High Added-value Chemicals – 85 Years of Lipase-based Enzymatic Catalysis. Part II
P. Borowiecki*
Journal of Polish Chemical Society (Wiadomości Chemiczne) 2015, 69, 391–430.
Lipases (EC 3.1.1.3; triacylglycerol acylhydrolases) are the most commonly used enzymes in biotransformations of organic compounds. In living organism lipases catalyze hydrolysis of higher fatty acid esters of glycerol, thus fulfill an essential function in metabolism of lipids (e.g. fats and oils) and lipoproteins. This year marks 125 years since J.R. Green has identified and described the first lipase isolated from germinated castor-oil beans (Ricinus communis L.) in the form of an extract showing hydrolytic properties. Plants, as well as bacteria are able to produce lipases what was reported in 1901 by Dutch scientist ‒ Christiaan Eijkman.
Lipases are also produced by fungi, yeasts, and various organs of higher organisms. A strong foundation, which had a huge impact on the development of global lipase-mediated biotransformations was the discovery made in 1935 and described in Biochemistry Journal and Biochemische Zeitschrift by Polish biochemist-enzymologist Ernest Alexander Sym (1893-1950) that these enzymes retain almost full catalytic activity even in nearly anhydrous organic solvents.
This was exactly fifty years before Russian chemist Alexander Klibanov in 1985 described a lipase-catalyzed reaction carried out in organic solvents. Since that moment, lipases have become extremely popular in both academic and industrial usage, nowadays being the most important among all biocatalysts used in biochemical processes carried out on an industrial scale.
The purpose of this article is to provide a brief characterization of the two most widely used in industrial biotransformations lipases ‒ lipase B from Candida antarctica (CAL-B) and lipase from Burkholderia cepacia (BCL) ‒ and familiarize the readers with the issues of biotechnological processes catalyzed by them. The specifics of a range of industrial applications based on lipase catalysis, including the chemical, pharmaceutical, cosmetic and food industries are also discussed.
11. Industrial Applications of Lipases in the Synthesis of High Added-value Chemicals – 85 Years of Lipase-based Enzymatic Catalysis. Part I
P. Borowiecki*
Journal of Polish Chemical Society (Wiadomości Chemiczne) 2015, 69, 431–464.
Biotransformations are processes, in which chemical reactions are catalyzed by isolated enzymes or whole cells containing them. Among the biocatalysts, lipases are the most commonly used chiral selectors that exhibit high chemo-, regio-, and stereo-selectivity toward wide spectrum of organic compounds of xenobiothic nature. Moreover, lipases are very stable and active in organic solvents, as well as in neat solvents or in supercritical fluids in the absence of added water.
Biotransformations by using lipases can be carried out at high substrate concentrations, at ambient temperature and neutral pH, without need for addition of cofactors, application of high pressures, extremely harsh reaction conditions or complex chemical apparatus. In addition, processes based on efficient biocatalytic technologies has proven to be beneficial for the chemical industry, as the lipases are able to catalyze reactions, which are not easily conducted by classical methods or in other cases allow reactions, which can replace several chemical steps.
The above-mentioned features of lipase-based biotransformations often cause significant improvement in energy efficiency (savings), and lead to a reduction in waste generation thereby making manufacturing processes even more economically attractive and environmentally acceptable. Since the mid-1980s the use of biotransformations with lipases in industry for the production of high added-value compounds, including pharmaceuticals, vitamins, cosmetics, fragrances and flavors, diagnostic preparations and therapeutics, high-tonnage preparation of agrochemicals, modified foods, nutraceuticals, detergents, polymers, advanced materials and biofuels has steadily increased.
In this part of the review article on industrial applications of lipases, next group of popularly utilized enzymes relevant for the production of high added-value chemicals are described. It was also shown on several examples that enzymatic catalysis can significantly simplify manufacturing processes of complex structures being green and economical alternative for conventional chemical-based processes.
10. Ionic Liquids and Potential Areas of their Applications in Chemical Industry
P. Borowiecki,* M. Bretner, J. Plenkiewicz
Journal of Polish Chemical Society (Wiadomości Chemiczne) 2015, 69, 271–296.
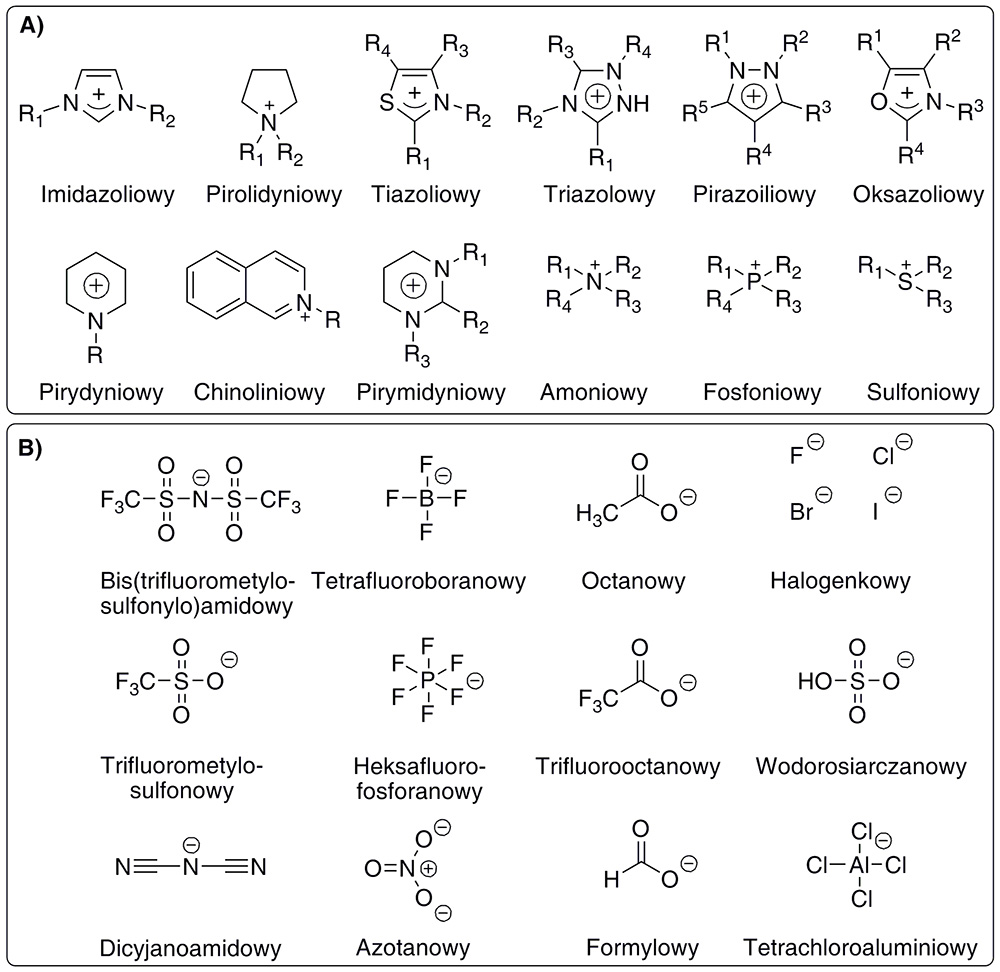
Ionic liquids (ILs) compose a group of chemical compounds of ionic nature consisting of organic cation and inorganic or organic anion, whose melting point does not exceed 100 °C (373.15 K). These compounds play a very important role in scientific investigations as well as in industrial organic synthesis, both in high-tonnage productions as well as low-tonnage technologies of high added-value chemicals and materials. Ionic liquids are mostly used as alternative media and/or catalysts for desired chemical reactions.
Multifunctionality and popularity of ionic liquids applications mainly stems from their beneficial physicochemical properties, such as: (i) very low vapor pressure, (ii) negligible viscosity, (iii) high overall- and thermal-stability, (iv) incombustibility and non-explosiveness, (v) high heat capacity, (vi) good solubility of most organic compounds (including polymers) as well as organometallic and inorganic compounds (including gases), (vii) low compressibility, and (viii) high electrochemical conductivity.
Moreover, ionic liquids (x) exist in liquid state in a very wide range of temperature, (xi) exhibit a wide range of electrochemical stability as well as (xii) improve properties of enzymes and other biocatalysts, positively impacting on their activity, stability, enantioselectivity and/or stereoselectivity. It is worth noting that ionic liquids increasingly constitute a target product of defined commercial characteristics, such in the case of: electrochemicals, chemotherapeutics, environmental anti-degradation agents, effective and safe agrochemicals etc. In this review, with 238 refs., development trends and potential applications of ionic liquids is presented.
2014
9. First Chemoenzymatic Stereodivergent Synthesis of both Enantiomers of Promethazine and Ethopropazine
P. Borowiecki,* D. Paprocki, M. Dranka
Beilstein Journal of Organic Chemistry 2014, 10, 3038–3055.
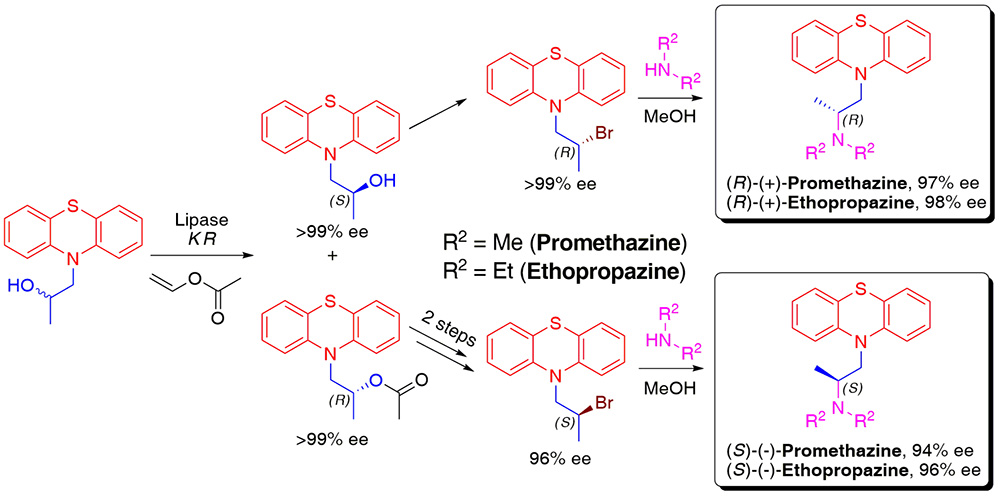
Enantioenriched promethazine and ethopropazine were synthesized through a simple and straightforward four-step chemoenzymatic route. The central chiral building block, 1-(10H-phenothiazin-10-yl)propan-2-ol, was obtained via lipase-mediated kinetic resolution protocol, which furnished both enantiomeric forms, with superb enantioselectivity (up to E = 844), from the racemate.
Novozym 435 and Lipozyme TL IM have been found as ideal biocatalysts for preparation of highly enantioenriched phenothiazolic-alcohols (up to >99% ee), which absolute configurations were assigned by Mosher’s methodology and unambiguously confirmed by XRD analysis. The obtained key-intermediates were further transformed into bromide derivatives by means of PBr3, and subsequently reacted with appropriate amine providing desired pharmacologically valuable (R)- and (S)-stereoisomers of title drugs in 84–98% ee range, respectively.
The modular amination procedure is based on solvent-dependent stereodivergent transformation of bromo derivative, which conducted in toluene gives mainly product of single inversion, whereas carried out in methanol provides exclusively the product of net retention. Enantiomeric excess of optically active promethazine and ethopropazine were established by HPLC measurements with chiral columns.
doi: 10.3762/bjoc.10.322
8. Synthesis of Novel Chiral TBBt Derivatives with Hydroxyl Moiety. Studies on Inhibition of Human Protein Kinase CK2α and Cytotoxicity Properties
P. Borowiecki,* A. M. Wawro, P. Wińska, M. Wielechowska, M. Bretner
European Journal of Medicinal Chemistry 2014, 84, 364–374.
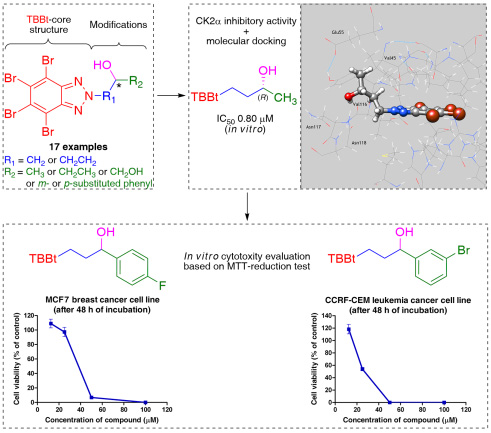
The efficient method for the synthesis of novel 4,5,6,7-tetrabromo-1H-benzotriazole (TBBt) derivatives bearing a single stereogenic center has been developed. New compounds with a variety of substituents at the meta– and para-position of the phenyl ring are reported. All of the presented compounds were obtained using classical synthetic methods, such as bromination of benzotriazole, and its subsequent alkylation by monotosylated arylpropane-1,3-diols, which in turn have been synthesized through reduction of the corresponding prochiral β-keto esters, and the selective monotosylation of the primary hydroxyl group.
The influence of the new and previously reported N-hydroxyalkyl TBBt derivatives on the activity of human protein kinase CK2α catalytic subunit was examined. The most active were derivatives with N-hydroxyalkyl substituents (IC50 in 0.80–7.35 µM range). A binding mode of (R)-1-(4,5,6,7-tetrabromo-2H-benzotriazol-2-yl)butan-3-ol (7b) to hCK2α has been proposed based on in silico docking studies.
Additionally, MTT-based cytotoxicity tests demonstrated high activities of novel 1-aryl-3-TBBt-propan-1-ol and 3-TBBt-propan-1,2-diol derivatives against human peripheral blood T lymphoblast (CCRF-CEM), and moderate anti-tumor activities against human breast adenocarcinoma (MCF7) cell lines.
7. Asymmetric Reduction of 1-(Benzoazole-2-ylsulfanyl)propan-2-ones using Whole Cells of Mortierella isabellina, Debaryomyces hansenii, Geotrichum candidum and Zygosaccharomyces rouxii
P. Borowiecki, M. Włoczewska, Z. Ochal*
Journal of Molecular Catalysis B: Enzymatic 2014, 109, 9–16.
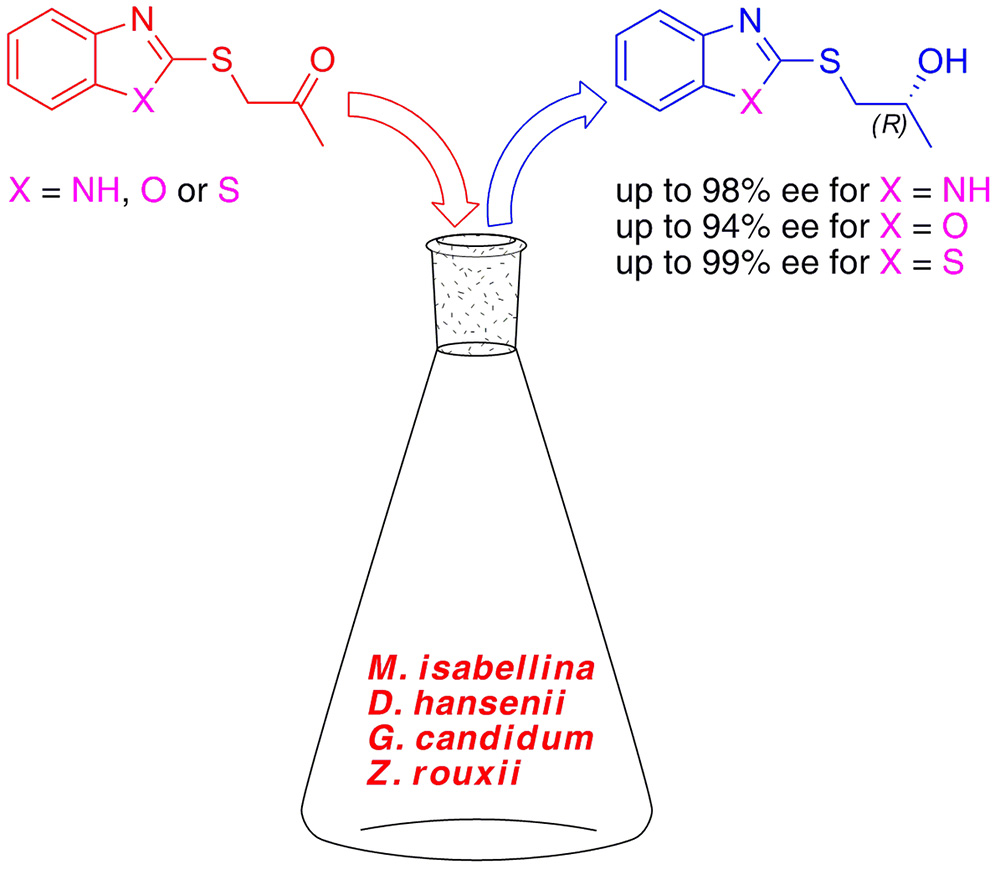
Growing cells of four fungal strains were used in reduction of 1-(benzoazol-2-ylsulfanyl)propan-2-ones 3a–c to corresponding (R)-(+)-1-(benzoazol-2-ylsulfanyl)propan-2-ols (R)-(+)-4a–c. All the investigated yeast strains displayed a very high activity toward prochiral ketones 3a–c converting them to the desired alcohols after relatively short reaction time (1–3.5 h).
The biotransformation products were isolated with moderate to good yields (45-89%) and in highly enantioenriched forms (94–99% ee). Stereoselective bioreduction of 1-(1H-benzimidazol-2-ylsulfanyl)propan-2-one (3a) by D. hansenii DSM 3428 growing cells provided the respective (R)-alcohol with >99% yield, in reasonable 65% isolated yield and in a highly stereoselective manner (98% ee) after 3 h of cultivation.
In the same culture, bioreduction of 1-(1,3-benzoxazol-2-ylsulfanyl)propan-2-one (3b) led to a 76% yield, 65% isolated yield and very high 94% ee of the formed (R)-alcohol. Similar 1.5 hour incubation of 1-(1,3-benzothiazol-2-ylsulfanyl)propan-2-one (3c) in G. candidum LOCK 105 culture resulted in the corresponding (R)-alcohol preparation in moderate 45% isolated yield with excellent enantiomeric purity (99% ee).
2013
6. Synthesis and Antimicrobial Activity of Imidazolium and Triazolium Chiral Ionic Liquids
P. Borowiecki,* M. Milner-Krawczyk, D. Brzezińska, M. Wielechowska, J. Plenkiewicz
European Journal of Organic Chemistry 2013, 4, 712–720.
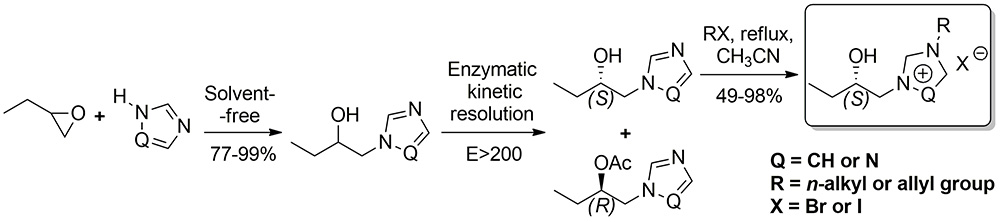
A simple and efficient procedure for the synthesis of new optically active imidazolium and triazolium ionic liquids in a three-step reaction sequence is described. In the first step, the ring opening of 1,2‐butylene oxide by imidazole or 1,2,4‐triazole resulted in the formation of N‐2‐hydroxybutylimidazole and N‐2‐hydroxybutyl‐1,2,4‐triazole, respectively.
In the second step, racemic mixtures of the secondary alcohols were resolved with good enantioselectivity by a lipase‐catalyzed transesterification with enol esters (vinyl acetate and 1‐acetoxy‐2‐methylcyclohexene).
In the final step, the optically active intermediates were alkylated with several haloalkanes to yield optically active ionic liquids. The inhibitory activity of the synthesized ionic liquids was tested towards gram‐negative and gram‐positive bacteria and fungi.
5. Chemoenzymatic Synthesis and Biological Evaluation of Enantiomerically Enriched 1-(β-Hydroxypropyl) Imidazolium- and Triazolium-based Ionic Liquids
P. Borowiecki,* M. Milner-Krawczyk, J. Plenkiewicz
Beilstein J. Org. Chem. 2013, 9, 516–525.
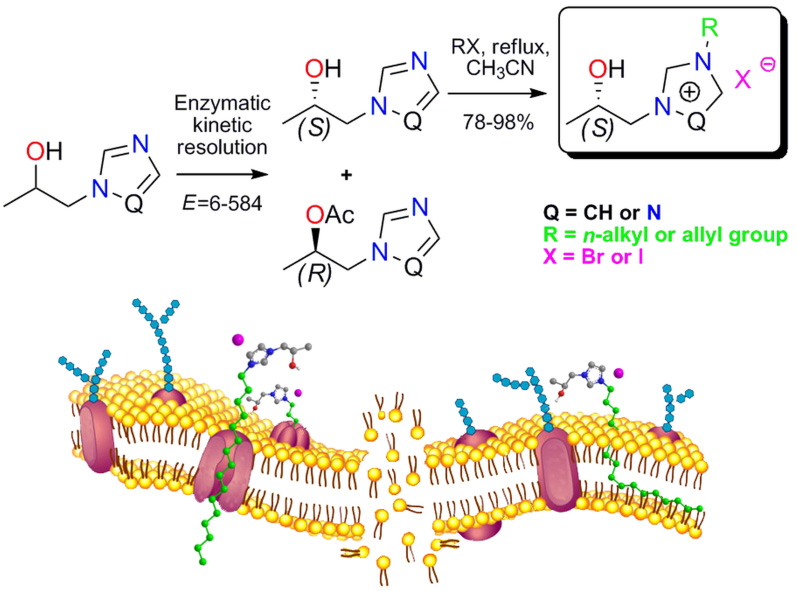
Racemic 1-(β-hydroxypropyl)azoles were prepared by solvent-free direct regioselective ring opening of 1,2-propylene oxide with imidazole or 1,2,4-triazole. Lipase-catalyzed transesterification of alcohols with vinyl acetate resulted in kinetic enantiomers resolution. Separated (S)-enantiomers of (+)-1-(1H-imidazol-1-yl)propan-2-ol and (+)-1-(1H-1,2,4-triazol-1-yl)propan-2-ol were quaternized with alkyl bromides or iodides, yielding novel optically active ionic liquids. Racemic salts were tested against a wide range of microorganisms.
doi: 10.3762/bjoc.9.56
4. Lipase-catalyzed Kinetic Resolution of 1-(1,3-Benzothiazol-2-ylsulfanyl)propan-2-ol with Antifungal Activity: A Comparative Study of Transesterification Versus Hydrolysis
P. Borowiecki, M. Fabisiak, Z. Ochal*
Tetrahedron 2013, 69, 4597–4602.
A study of chemoenzymatic synthesis of both enantiomers of 1-(1,3-benzothiazol-2-ylsulfanyl)propan-2-ol was carried out. Several commercially available lipase preparations were tested as biocatalysts in the kinetic resolution process of target compound by enantioselective transesterification and/or hydrolysis. CAL-B (Novozym 435) was found to be the optimal catalyst.
The lipase-mediated hydrolysis approach appeared to be superior to the transesterification reaction. Absolute configuration of the obtained alcohol was postulated, applying modified Mosher’s methodology. The inhibitory activity of the synthesized benzothiazole derivatives against pathogenic fungi was checked.
3. First Chemoenzymatic Synthesis of (R)- and (S)-1-(9H-Fluoren-9-yl)ethanol
P. Borowiecki, S. Balter, I. Justyniak, Z. Ochal*
Tetrahedron: Asymmetry 2013, 24, 1120–1126.
A simple chemoenzymatic synthesis of 1-(9H-fluoren-9-yl)ethanol stereoisomers is described. The enantiomers were resolved by a kinetically controlled transesterification with vinyl acetate in the presence of commercially available lipases. High-throughput screening and subsequent exhaustive investigation of the utility of the lipases in a stereoselective process of introducing chirality have been carried out.
Lipase A from Candida antarctica as a cross-linked aggregate (CAL-A-CLEA) was the most efficient enzyme for the resolution of the title compound providing (S)-1-(9H-fluoren-9-yl)ethanol and its (R)-acetate in enantiopure form (>99% ee). Under mild reaction conditions, excellent enantioselectivity (E = 407), and good isolated yields of the products were obtained.
2. Studies on the Chemoenzymatic Synthesis of (R)- and (S)-Methyl 3-Aryl-3-hydroxypropionates: The Influence of Toluene Pretreatment of Lipase Preparations on Enantioselective Transesterifications
P. Borowiecki,* M. Bretner
Tetrahedron: Asymmetry 2013, 24, 925–936.
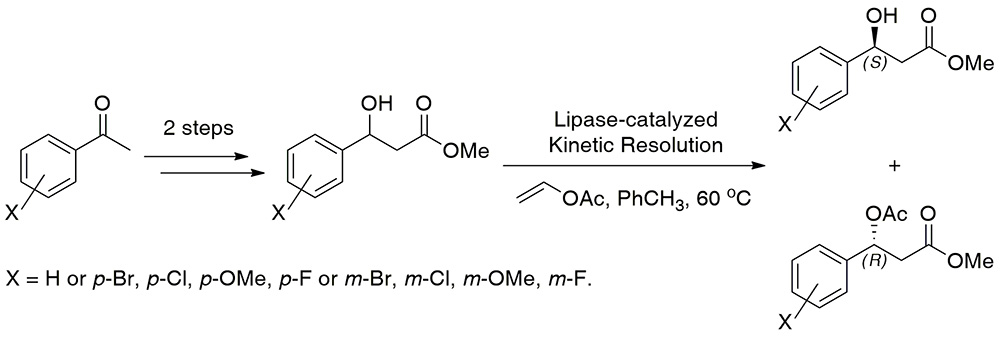
Two series (para– and meta-substituted) of racemic methyl esters of 3-aryl-3-hydroxypropionic acid were prepared after which the enantiomers were separated by an enzyme-catalyzed transesterification. Several lipases were investigated as the catalyst.
The influence of the enzyme pretreatment, as well as substrate concentration, reaction temperature, stirring manner, and substrate conversion on the stereochemical outcome of the biotransformation process were investigated in detail.
The best results were achieved by using solvent-pretreated lipase from Pseudomonas fluorescens or Burkholderia cepacia suspended in toluene, and vinyl acetate as the acetyl group donor.
2012
1. Preparation and Thermal Stability of Optically Active 1,2,4-Triazolium-based Ionic Liquids
P. Borowiecki,* M. Poterała, J. Maurin, M. Wielechowska, J. Plenkiewicz
ARKIVOC 2012, (viii), 262–281.
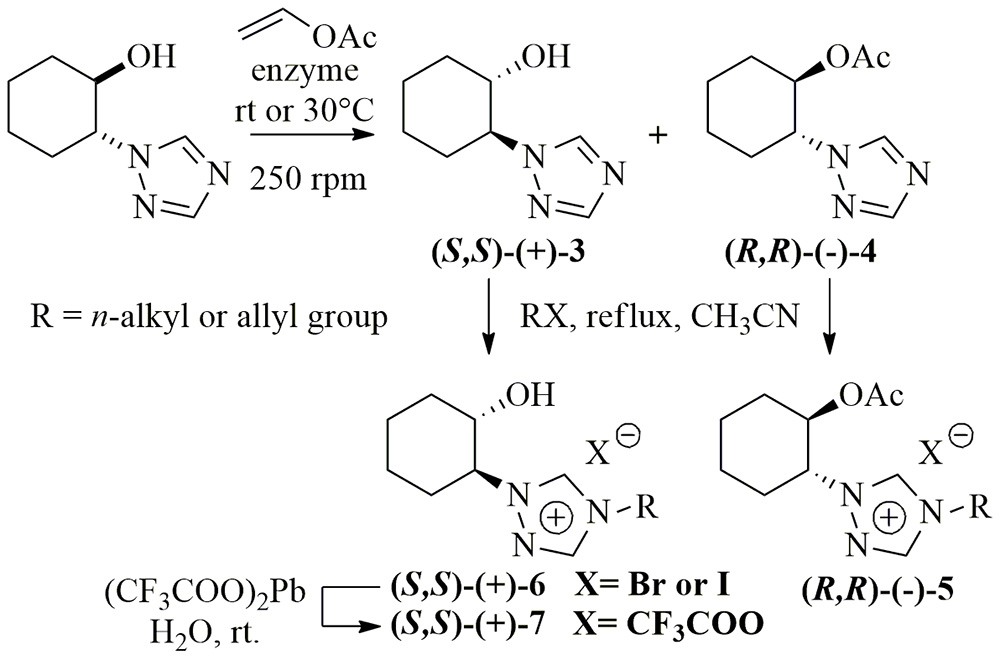
The synthesis of optically active ionic liquids, in a four-step reaction sequence, is described. In the first step an oxirane ring of cyclohexene oxide was opened with 1,2,4-triazole, yielding a racemic mixture of (1R,2R)- and (1S,2S)-2-(1H-1,2,4-triazol-1-yl)cyclohexanol.
Kinetic resolution of the racemate by a lipase catalyzed transesterification with vinyl acetate followed by alkylation (quaternization) of the triazole ring resulted in the appropriate optically active salts formation. After the anion metathesis, thermally stable novel chiral ionic liquids were obtained.